Avicenna Journal of Clinical Microbiology and Infection. 12(3):139-151.
doi: 10.34172/ajcmi.3692
Original Article
Harnessing the Power of Bioinformatics Pipelines to Develop an Anti-SARS-CoV-2 Pan-Species Vaccine: A Post-Pandemic Description
Mohammad Roayaei Ardakani Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing, 1 
Fatemeh Yaghoobizadeh Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing, 1, *
Ghader Bashiri Conceptualization, Writing – review & editing, 2
Mohammad Mehdi Ranjbar Conceptualization, Writing – review & editing, 3
Author information:
1Department of Biology, Faculty of Science, Shahid Chamran University of Ahvaz, Ahvaz, Khouzestan, Iran
2Science School of Biological Sciences, The University of Auckland, Auckland, New Zealand
3Razi Vaccine and Serum Research Institute, Karaj, Alborz, Iran
Abstract
Background: The emergence of infectious agents enforces rapid prophylactic/therapeutic development. Accordingly, multi-epitope vaccines represent a milestone in the second-generation vaccines to break the SARS-CoV-2 transmission chain, a zoonotic virus, and decrease long coronavirus (COVID-19) complications. Thus, this study aimed to present a potent anti-SARS-CoV-2 pan-species vaccine candidate via computational workflow to address this hypothesis: Could a multi-epitope construct provoke effective anti-SARS-CoV-2 protection as a bioinformatics solution?
Methods: The B-cell and T-cell epitopes of the S1 domain, N, Nsp4, Nsp12, and ORF6 proteins were predicted regarding the significant role of viral proteins in the infection cycle. Then, the final construct was designed as (1) scaffolding by the top-ranked epitopes, β-defensin, His6-tag, (2) structural modeling/refinement, (3) molecular docking/dynamics simulation, and (4) validation.
Results: The 15 linear B-cell, 7 helper T-cell, and 11 cytotoxic T-cell epitopes were embedded within the construct, all located outside the hyper-variable regions of target proteins. Following the quality assessment, its best binding affinity was observed with TLR4, TLR8, HLA-DRB1*01:01, HLA-A*03:01, and ACE2. The strong interactions were also observed with a validated single-chain variable fragment as a preliminary in silico indication about its possible in vivo efficiency. Moreover, the messenger RNA stability and robust immune stimulation were verified. Not only was non-significant homology found between the multi-epitope vaccine construct and some viral hosts/reservoir proteomes, but also significant coverage was detected between human immune receptors and their counterparts in domestic, wildlife, and captive animals. Therefore, it could be a sign of the possible efficiency of the construct as a pan-species vaccine.
Conclusion: Despite the end of the COVID-19 pandemic, a combinatorial workflow was introduced to design a vaccine with potential application in various species, whereas many other studies have not employed all of these criteria, and according to our information, this is one of the few works to introduce such constructs.
Keywords: In silico analysis, Multi-epitope vaccines, SARS-CoV-2, Vaccine design, Zoonotic viruses, Pan-species vaccines
Copyright and License Information
© 2025 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Please cite this article as follows: Roayaei Ardakani M, Yaghoobizadeh F, Bashiri G, Ranjbar MM. Harnessing the power of bioinformatics pipelines to develop an anti-SARS-CoV-2 pan-species vaccine: a post-pandemic description. Avicenna J Clin Microbiol Infect. 2025;12(3):139-51. doi:10.34172/ajcmi.3692
Introduction
The SARS-CoV-2, the causative agent of the most recent pandemic, belongs to the Betacoronavirus genus, Orthocoronavirinae subfamily, Coronaviridae family, order Nidovirales. It was nominated by the International Committee on Taxonomy of Viruses based on the phylogenetic relationship with human coronaviruses and SARS-related bat coronaviruses. On the other hand, other mammals, avians, and domestics are also susceptible to this virus due to its zoonotic nature. Thus, it could pose a challenge to the veterinary profession and cause economic loss (1-3).
The ~30 kbp, single-stranded RNA genome of this enveloped virus contains at least 10 open reading frames (ORFs) that encode 4 structural proteins (spike [S], membrane [M], nucleocapsid [N], and envelope [E]), 16 non-structural proteins (Nsp1-Nsp16), and several accessory proteins, including ORF3a, 3b, 6, 7a, 7b, 8, 9b, and ORF10 (3-8).
The SARS-CoV-2 infectivity initiates via binding to the permissive host cells mediated by two main receptors, that is, the angiotensin-converting enzyme 2 (ACE2) and the cell surface-associated transmembrane serine protease 2 (TMPRSS2). Different parts of the spike glycoprotein, as a highly viral immunogen, are involved in these interactions. Among them, the receptor-binding domain (RBD) of the S1 domain makes strong interactions with the ACE2, thereby playing an important role in host cell tropism and viral entry. This is followed by the dissociation of N phosphoprotein from the genome and the release of the positive-sense genomic RNA into the cytoplasm to initiate the infectious cycle. The N protein facilitates the assembly of viral particles and the modulation of the host immune responses as well (3,4,9-14).
The SARS-CoV-2 employs a discontinuous replication strategy through the formation of double-membrane vesicles (DMVs), a replication-transcription complex, and similar processes, achieved by viral proteins’ cooperation. For example, Nsp3/PLPro and Nsp5/3CLPro/MPro proteases mediate the autoproteolytic processes, while Nsp4, Nsp3, and Nsp6 facilitate DMV formation. Thereafter, the RNA genome is replicated by Nsp12, an RNA-dependent RNA-polymerase (RdRp), within this DMV. Subsequently, the nascent sub-genomic RNAs are translated into structural and accessory proteins. Additionally, SARS-CoV-2 employs immune modulators/evaders, such as ORF6, to impair the host immune defense (9,15).
Two important issues exist about SARS-CoV-2, namely, (1) variations in viral severity by the emergence of various variants-of-interest, variants-under-monitoring, and variants-of-concern and (2) many complications considering the long coronavirus (COVID-19) or post-COVID-19 syndrome with the potential impact on the patient’s quality of life (16,17). Both of these issues highlight the need for developing a novel and efficient vaccine with potent efficiency (7,18-20). Accordingly, this work aims to resolve the following questions:
Could the in silico/computational studies make a basis to design an efficient anti-SARS-CoV-2 multi-epitope vaccine construct (MEV)? Does this vaccine have an in silico pan-species activity to cope with the viral zoonotic nature?
Therefore, an in-depth bioinformatics/computational analysis is conducted to formulate a novel pan-species vaccine and evaluate its in silico efficiency.
Material and Methods
Protein Sequence Retrieval
The AA sequences of target proteins were downloaded from the National Center for Biotechnology (NCBI) database (https://www.ncbi.nlm.nih.gov/) in FASTA format with the following accession numbers:
S1 domain (YP_009724390.1), N (YP_009724397.2), ORF6 (YP_009724394.1), Nsp4 (YP_009725300.1), and Nsp12/RdRp (YP_009725307.1) (5,13,15,21,22).
Conservative/Variable Regions Identification
A dataset of ~200 sequences was retrieved from the NCBI virus database (https://www.ncbi.nlm.nih.gov/labs/virus/) from variants of concern and variants of interest for each protein. Then, the multiple sequence alignment and Shannon entropy (Hx) plot analysis were performed to find the conserved domains using BioEdit, version 7.0.9.0 (4,13).
Epitope Mapping
Linear B-Cell Epitope Prediction
Vaccine constructs require potent B-cell-stimulating epitopes to induce humoral immunity. Accordingly, potential LBLs were predicted by ABCpred (https://webs.iiitd.edu.in/raghava/abcpred/ABC_method.html), BCPred (http://crdd.osdd.net/raghava/bcepred/), and IEDB (http://tools.iedb.org/bcell/) servers. Top-ranked and non-overlapped epitopes were selected (3,4,13,21,23-25).
T-Cell Epitope Prediction
T-cell epitopes could interact with major histocompatibility complexes (MHCs) I and II to trigger downstream responses (6). Hence, the cytotoxic T-cell or TC (CTL) and helper T-cell or TH (HTL) epitopes were predicted by the NetMHC I (https://services.healthtech.dtu.dk/services/NetMHC-4.0/) and NetMHC II (https://services.healthtech.dtu.dk/services/NetMHCII-2.3/) servers, respectively. Epitopes with half-maximal inhibitory concentration ≤ 500 nM were refined by IEDB tools (4,5,6,13,19,21,23,25-27,28).
Immunogenicity Profiling
The selected epitopes were further refined according to their possible antigenicity (VaxiJen: https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html), toxicity (Toxinpred: https://webs.iiitd.edu.in/raghava/toxinpred/algo.php), and allergenicity (AllerTop: https://www.ddg-pharmfac.net/AllerTOP/). The cytokine-inducing ability was also used for epitope refinement by interferon gamma (IFNγ) (http://crdd.osdd.net/raghava/ifnepitope/), interleukin (IL)-4 (http://crdd.osdd.net/raghava/il4pred/), and IL-10 (http://crdd.osdd.net/raghava/IL-10pred/) servers. The TepiTool (http://tools.iedb.org/tepitool/) was utilized to predict the potential immune tolerance of CTL/HTL epitopes by T regulatory cells. It should be noted that a higher percentile rank is better (5,13,21,23-25,27,29).
Epitope Conservancy Analysis
To ensure vaccine efficacy against diverse SARS-CoV-2 strains, the shortlisted epitopes were evaluated using the IEDB conservancy analysis tool in the protein dataset (http://tools.iedb.org/conservancy/). Epitopes with a ≥ 95% score were selected (5,11,30,31).
Human Leukocyte Antigen Population Coverage Analysis
Considering the polymorphic nature of HLA alleles, population coverage analysis was performed for selected CTL and HTL epitopes using the IEDB population coverage tool (http://tools.iedb.org/population/) by default parameters, with special focus on global and regional (Iran) populations (4,5,23,31).
Multi-epitope Vaccine Construct Scaffolding
The MEV was engineered by connecting the prioritized epitopes with the three types of linkers to enhance antigen presentation, including AYY (alanine tyrosine tyrosine), GPGPG (glycine-proline rich), and KK (lysine-lysine). Two other elements were also included at the N-terminal and C-terminal of the vaccine construct, respectively, namely, (1) human β‐defensin 3 (hβD-3; UniProt ID Q5U7J2), fused to the first CTL using an EAAAK (Glu-Ala-Ala-Ala-Lys) linker to boost immunogenicity and (2) 6xHis-tag for the next purification purposes (4,5,19,25,27,29,32).
Physicochemical and Solubility Characterization
The vaccine construct properties were evaluated using ProtParam, ExPASy server (https://web.expasy.org/protparam/). Antigenicity, allergenicity, and solubility were also predicted by VaxiJen v2.0, ANTIGENpro (http://scratch.proteomics.ics.uci.edu/), AllergenFP (version 1.0; http://ddg-pharmfac.net/AllergenFP/), AllerTop (version 2.0), SOLpro (https://scratch.proteomics.ics.uci.edu/explanation.html#SOLpro), and SoluProt (https://loschmidt.chemi.muni.cz/soluprot/) servers. Additionally, the epitope masking potential of the construct was assessed based on the prediction of potential N-glycosylation sites by NetNGlyc (version 1.0; http://cbs.dtu.dk/services/NetNGly/) (4,13,25-27,31,32).
Cross-Reactivity Evaluation
The MEV was aligned against the human proteome by the Blastp tool, the NCBI server, to minimize potential autoimmune responses. The search parameters were restricted to Homo sapiens (taxid 9606). Moreover, the cross-reactivity was also assessed against the following taxa to get a preliminary computational insight into the possible pan-species capacity of the MEV:
Macaca (taxid 9539), gorilla (taxid 9592), lion (taxid 9689), tiger (taxid 9694), white-tailed deer (taxid 9874), mink (taxid 9666), domestic ferret (taxid 9669), cattle (taxid 9903), dog (taxid 9615), cat (taxid 9685), and spotted hyena (taxid 9678) (4,13,26).
Structure Modelling
The PSIPRED 4.0 (http://bioinf.cs.ucl.ac.uk/psipred/) and I-TASSER (https://zhanggroup.org/I-TASSER/) servers were applied to determine the 2-dimensional (2D) and 3D structures of the construct, respectively (13,24,25,27,29,31).
Three-Dimensional Structure Refinement and Validation
Following the structure refinement by GalaxyRefine (http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE), the best model was selected based on the root-mean-square deviation (RMSD), clash scores, high-accuracy global distance test, MolProbity, rotamer quality, and Ramachandran plot analysis. Further validations were performed using SAVES (version 6.0; https://saves.mbi.ucla.edu/) and proSA-web (https://prosa.services.came.sbg.ac.at/prosa.php) servers to assess energetically favored/unfavored psi (ψ) and phi (φ) angles of amino acid residues, and the potential structural errors, respectively (4,8,13,23,25,27,29,33).
Conformational B-Cell Epitope Prediction
A critical aspect of vaccine design is the prediction of conformational BCEs, which arise from protein folding. Accordingly, the MEV conformational epitopes were predicted by the Ellipro server (http://tools.iedb.rg/ellipro/) (5,26,27,33).
Protein-Protein Docking
Strong interactions between vaccine-immune receptors (e.g., toll-like receptors [TLRs], MHC I and II) are necessary to induce an effective immune response. Thus, the HEX 8.0.0 was used to perform protein-protein docking and assess the binding interactions (5,19). The structures of the following immune receptors were retrieved from the Protein Data Bank (PDB) (https://www.rcsb.org/):
TRL2 (PDB ID 2Z7X), TLR3 (PDB ID 5GS0), TLR4 (PDB ID 4G8A), TLR5 (PDB ID 3J0A), TLR7 (PDB ID 7CYN), TLR8 (PDB ID 4QC0), HLA-A*03:01 (PDB ID 7L1C), HLA-A*11:01 (PDB ID 8WTE), HLA-A*68:01 (PDB ID 6PBH), HLA-B*35:01 (PDB ID 4PRN), HLA-DRB1*01:01 (PDB ID 1AQD), and HLA-DRB1*09:01 (PDB ID 1D5Z)
The MEV and ACE2 (PDB ID 6M0J) interactions were assessed as well. Additionally, the possible pan-species activity of MEV was assayed based on the coverage score between human TLRs, MHC I/II, and the studied viral host/reservoirs via the blastp running.
Pre-docking processing was performed by the DockPrep tool, UCSF ChimeraX 1.1, and the RMSD was calculated to validate the docking accuracy. Binding energy (ΔG in kcal/mol) and the dissociation constant (Kd) were also estimated using the PRODIGY tool (https://rascar.science.uu.nl/prodigy/) (4,18,19,29,34-36).
An In Silico Indication of the Possible In Vivo Efficiency
The molecular docking was also performed with a validated single-chain variable fragment (scFv). This anti-RBD-scFv was designed, expressed in the Escherichia coli BL21, and purified elsewhere, and its efficiency was confirmed by experimental tests, such as sandwich-enzyme-linked immunosorbent assay (3). As a result, this interaction might be a preliminary in silico indication about its possible in vivo efficiency to make a foundation for further experimental assays.
Molecular Dynamics Simulation
The CABS-flex 2.0 (https://biocomp.chem.uw.edu.pl/CABSflex2) and iMODS (https://imods.iqf.csic.es/) servers were used to analyze the molecular dynamicssimulations of docked complexes according to dynamic behavior, binding mode, and conformational flexibility (8,35,37-40).
Codon Optimization and In Silico Cloning
The finalized DNA sequence of the construct was prepared as (1) back-translation of the AA sequence to its possible DNA sequence by the backtranseq tool (https://www.ebi.ac.uk/jdispatcher/st/emboss_backtranseq) and (2) the codon adaptive index (CAI) calculation by the GenSmart codon optimization tool (https://www.genscript.com/gensmart-free-gene-codon-optimization.html). It was also prepared as the (3) stop codon (TAA) insertion at the 3’ end and (4) the insertion of NcoI (C↓CATGG) and BamHI (G↓GATCC) sequences at the 5’/N and 3’/N ends, respectively (6,19). Following the in silico cloning of this construct into pET28( + ), the proper cloning process was confirmed by gel electrophoresis simulation via SnapGene® (https://www.snapgene.com) (4,6,19).
Multi-Epitope Vaccine Expression Prediction
The stability of messenger RNA (mRNA) secondary structure and its expression are directly correlated. Accordingly, the RNAfold (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) and UNAfold (http://www.unafold.org/mfold.php) servers were applied to perform this analysis by default parameters (5,21,27).
In Silico Immune Response Analysis
The immunogenic behavior of the designed vaccine was profiled by the C-ImmSim (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php) server with the four antigen injections at four-week intervals. The risk of cytokine storm was also estimated by calculating the Simpson’s diversity index (D) (4,13,29,37,40).
Results
Conservative/Variable Region Analysis
Based on the Shannon entropy (Hx) plot (Figure 1), the 11, 65, 139, 212, 344, 376, 422, 457, and 489 positions of the S1 protein were identified as the hyper-variable regions with scores greater than the threshold of 1.0, whereas position 503 showed a borderline variation. In contrast, no hyper-variable regions were detected in other proteins. This indicates a high conservation among various variants, ensuring the vaccine targets the conserved regions of SARS-CoV-2 variants less prone to mutation (4,13).
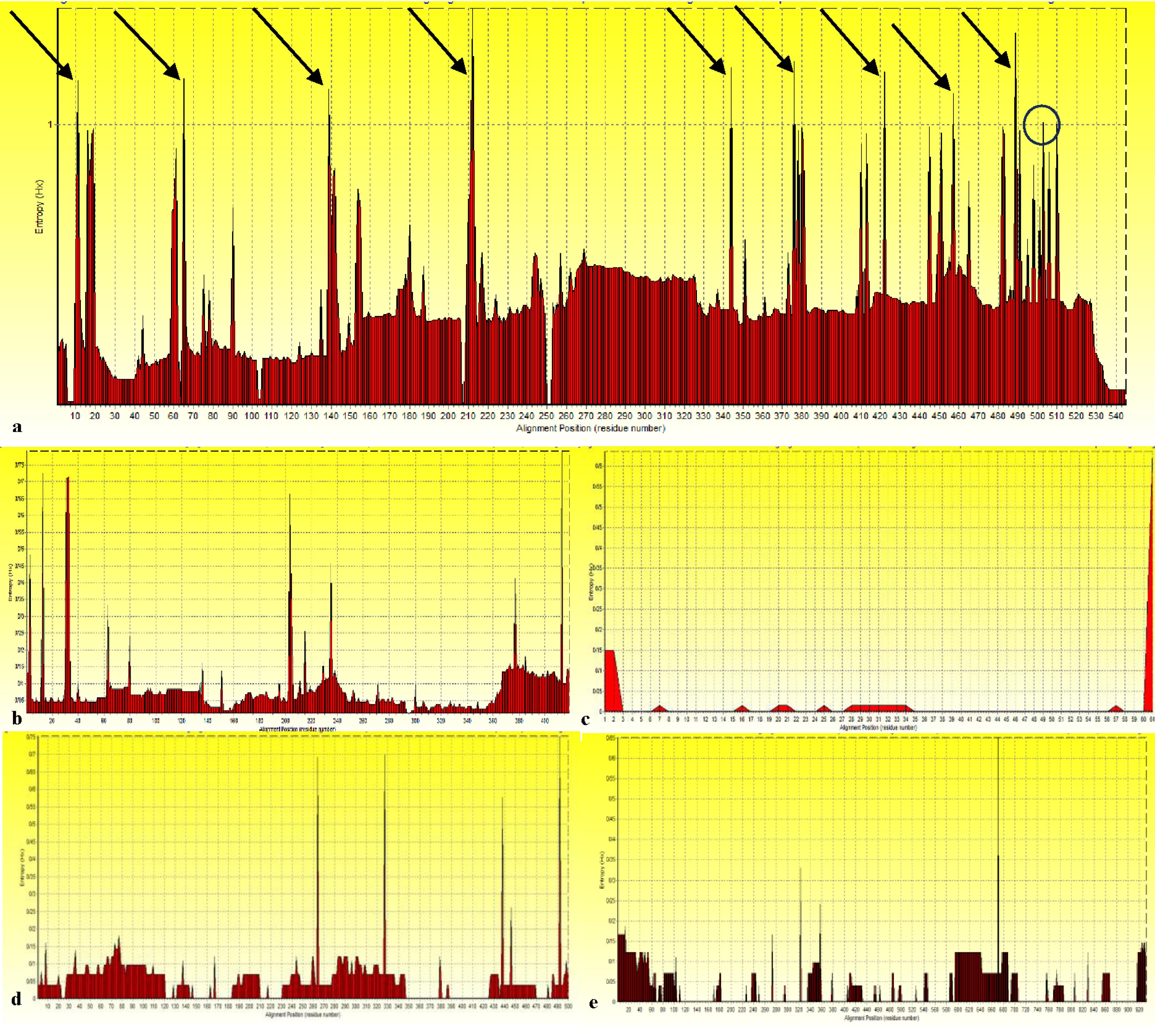
Figure 1.
Assessment of Variability in Protein Sequences by Entropy Plot (Hx): (a) S1 domain, (b) N protein, (c) ORF6 Protein, (d) Nsp4 Protein, and (e) Nsp12 Protein. Note. HVR: Hyper-variable region; ORF: Open reading frame; Nsp: Nonstructural protein. Arrows indicate HVRs with a score greater than the threshold (1.0); the borderline variation is shown with circles
.
Assessment of Variability in Protein Sequences by Entropy Plot (Hx): (a) S1 domain, (b) N protein, (c) ORF6 Protein, (d) Nsp4 Protein, and (e) Nsp12 Protein. Note. HVR: Hyper-variable region; ORF: Open reading frame; Nsp: Nonstructural protein. Arrows indicate HVRs with a score greater than the threshold (1.0); the borderline variation is shown with circles
Epitope Mapping
B-Lymphocyte Epitope Prediction
BCEs trigger a humoral immunity through the B-cell stimulation to produce various immunoglobulins and interact with other immune mediators, such as TH cells. Hence, the 3, 2, 2, 6, and 2 potent epitopes were identified in S1, N, Nsp4, Nsp12, and ORF6 sequences, respectively (Table 1).
Table 1.
Final Set of Top-Ranked LBL Epitopes
|
Epitope
|
Position
|
Antigenicity Score (Cut off Score, 0.4)
|
Allergenicity
|
Toxicity
|
IFNγ
|
IL-4
|
IL-10
|
Conservancy
|
Mean Entropy Score
|
| S1 domain |
| AAYYVGYL |
251-258 |
0.5218 |
Non-allergen |
Non-toxin |
Inducer (0.1649038) |
Inducer (0.32) |
Inducer (0.432) |
94.74% |
0.296 |
| KGIYQTSNFRVQPTES |
298-313 |
0.7664 |
Non-allergen |
Non-toxin |
Inducer (0.49561896) |
Inducer (0.44) |
Inducer (0.548) |
100.00% |
0.456 |
| NDLCFTNVYADSFVIR |
376-391 |
0.6806 |
Non-allergen |
Non-toxin |
Inducer (0.39757221) |
Inducer (0.23) |
Inducer (0.62) |
94.74% |
0.518 |
| N protein |
| KSAAEASKKPRQKRTA |
249-264 |
0.4636 |
Non-allergen |
Non-toxin |
Inducer (1.8035772) |
Inducer (0.36) |
Inducer (0.42800847) |
98.11% |
0.052 |
| RRGPEQTQGNFGDQ ELIRQGTDYK |
276-299 |
0.6277 |
Non-allergen |
Non-toxin |
Inducer (3.6135807) |
Inducer (1.08) |
Inducer (0.33673551) |
100.00% |
0.038 |
| Nsp4 protein |
| SAVGNICYTPSKLI |
127-140 |
0.9967 |
Non-allergen |
Non-toxin |
Inducer (1.8187864) |
Inducer (1.15) |
Inducer (0.49027266) |
97.73% |
0.033 |
| MDTTSYREAA |
458-467 |
0.5858 |
Non-allergen |
Non-toxin |
Inducer (1.7210174) |
Inducer (0.25) |
Inducer (0.31909215) |
100.00% |
0.039 |
| Nsp12 protein |
| ADLVYALRHFDEGNCD |
125-140 |
0.8534 |
Non-allergen |
Non-toxin |
Inducer (1.0) |
Inducer (1.28) |
Inducer (0.575) |
100.00% |
0.0 |
| TYHPNCVNC |
293-301 |
1.2280 |
Non-allergen |
Non-toxin |
Inducer (1.5703182) |
Inducer (0.28) |
Inducer (0.612) |
100.00% |
0.009 |
| VELKHFFF |
435-442 |
1.6097 |
Non-allergen |
Non-toxin |
Inducer (0.84217611) |
Inducer (0.28) |
Inducer (0.627) |
100.00% |
0.0 |
| ISAKNRA |
548-554 |
2.3024 |
Non-allergen |
Non-toxin |
Inducer (0.44859093) |
Inducer (0.27) |
Inducer (0.483) |
100.00% |
0.0 |
| GVSICSTMTNRQFHQK |
559-574 |
0.7298 |
Non-allergen |
Non-toxin |
Inducer (2.3251987) |
Inducer (1.05) |
Inducer (0.518) |
100.00% |
0.0 |
| NFKSVLYYQN |
781-790 |
0.8990 |
Non-allergen |
Non-toxin |
Inducer (1.6741401) |
Inducer (0.28) |
Inducer (0.54) |
100.00% |
0.024 |
| ORF6 protein |
| KVSIWNLDYIINLIIK |
23-38 |
0.5428 |
Non-allergen |
Non-toxin |
Inducer (0.98469288) |
Inducer (0.22) |
Inducer (0.548) |
96.55% |
0.008 |
| IKNLSKSLTENKYSQLD EEQ |
37-56 |
0.4801 |
Non-allergen |
Non-toxin |
Inducer (1.0438457) |
Inducer (0.55) |
Inducer (0.572) |
100.00% |
0.0 |
Note. LBL:Linear B-cell epitope; Nsp: Nonstructural protein; ORF: Open reading frame; IFNγ: Interferon gamma; IL: Interleukin.
T-Lymphocyte Epitope Prediction
Only 11 CTL (Table 2) and 7 HTL (Table 3) epitopes met the necessary criteria. It should be noted that data were unavailable in the Allele Frequency Net Database for the following MHC II-restricted alleles:
HLA-DRB3*01:01, HLA-DRB3*02:02, HLA-DRB4*01:01, HLA-DRB5*01:01, HLA-DPA1*01:03/DPB1*02:01, HLA-DPA1*02:01/DPB1*01:01, HLA-DPA1*01:03/DPB1*04:01, HLA-DPA1*02:01/DPB1*02:01, HLA-DPA1*03:01/DPB1*04:02, HLA-DPA1*02:01/DPB1*05:01, HLA-DQA1*01:01/DQB1*05:01, HLA-DQA1*05:01/DQB1*03:01, and HLA-DQA1*04:01/DQB1*04:02
Table 2.
Final Set of Top-Ranked CTL Epitopes
|
Epitope
|
Position
|
NetMHC I Percentile Rank
|
Supertype
|
MHC Class I Allele
|
IC50 (nM)
|
TepiTool Percentile Rank
|
Proteasome Score
|
TAP Score
|
Processing Score
|
Immunogenicity Score
|
Antigenicity Score (Cut off Score, 0.4)
|
Allergenicity
|
Toxicity
|
Conservancy
|
Mean entropy Score
|
Population Coverage
|
| S1 Domain |
| WTAGAAAYY |
246-255 |
0.02 |
A26, A1, A3, B62, B7 |
HLA-A*01:01 |
18.77 |
0.06 |
1.25 |
1.25 |
2.49 |
0.15259 |
0.6306 |
Non-allergen |
Non-toxin |
94.74% |
0.264 |
47.64% (42.71%) |
| HLA-A*26:01 |
9.27 |
0.03 |
| HLA-A*68:01 |
22.02 |
0.76 |
| HLA-A*30:02 |
25.11 |
0.18 |
| HLA-B*15:01 |
50.94 |
0.65 |
| HLA-B*35:01 |
60.67 |
0.39 |
| N Protein |
| LSPRWYFYY |
104-112 |
0.08 |
A1, A3, B58 |
HLA-A*01:01 |
95.14 |
0.29 |
1.22 |
1.29 |
2.51 |
0.35734 |
1.2832 |
Non-allergen |
Non-toxin |
100.00% |
0.075 |
42.63% (47.32%) |
| HLA-A*11:01 |
509.61 |
3.2 |
| HLA-A*30:02 |
44.28 |
0.52 |
| HLA-B*57:01 |
470.37 |
0.62 |
| HLA-B*58:01 |
299.94 |
0.79 |
| Nsp4 Protein |
| LAYYFMRFR |
297-305 |
1.00 |
A3 |
HLA-A*11:01 |
217.84 |
2.1 |
1.1 |
0.75 |
2.0 |
0.05586 |
0.5648 |
Non-allergen |
Non-toxin |
100.00% |
0.095 |
41.4% (48.16%) |
| HLA-A*31:01 |
10.12 |
0.31 |
| HLA-A*33:01 |
20.39 |
0.23 |
| HLA-A*68:01 |
9.51 |
0.61 |
| SVIYLYLTF |
343-351 |
0.25 |
A3, A24, B7, B62 |
HLA-A*23:01 |
75.99 |
0.25 |
1.47 |
1.17 |
2.64 |
0.03852 |
1.0482 |
Non-allergen |
Non-toxin |
100.00% |
0.031 |
49.21% (55.87%) |
| HLA-A*32:01 |
46.11 |
0.09 |
| HLA-A*24:02 |
483.89 |
0.52 |
| HLA-B*15:02 |
260 |
0.46 |
| LAHIQWMVM |
360-368 |
1.30 |
B7, B8 |
HLA-B*08:01 |
462.21 |
1.2 |
1.14 |
0.17 |
1.3 |
0.11605 |
1.3148 |
Non-allergen |
Non-toxin |
100.00% |
0.0 |
30.21% (56.94%) |
| HLA-B*35:01 |
135.73 |
0.81 |
| FTPLVPFWI |
369-377 |
0.16 |
A2, B62 |
HLA-A*02:01 |
30.76 |
2.7 |
1.06 |
0.17 |
1.23 |
0.24322 |
0.4253 |
Non-allergen |
Non-toxin |
100.00% |
0.0 |
60.33% (52.11%) |
| HLA-B*15:01 |
110.35 |
26 |
| Nsp12 Protein |
| ILHCANFNV |
307-315 |
0.09 |
A2 |
HLA-A*02:01 |
7.37 |
0.53 |
0.99 |
0.14 |
1.13 |
0.08328 |
0.5385 |
Non-allergen |
Non-toxin |
100.00% |
0.0 |
44.32% (41.48%) |
| HLA-A*02:03 |
8.83 |
0.71 |
| HLA-A*02:06 |
15.18 |
1.3 |
| VPFVVSTGY |
338-346 |
0.90 |
B7 |
HLA-B*35:01 |
11.43 |
0.01 |
1.25 |
1.2 |
2.45 |
0.01657 |
1.1641 |
Non-allergen |
Non-toxin |
100.00% |
0.076 |
19.85% (47.28%) |
| HLA-B*53:01 |
311.85 |
0.09 |
| KSAGFPFNK |
500-508 |
0.12 |
A3 |
HLA-A*03:01 |
22.93 |
0.02 |
0.87 |
0.26 |
1.13 |
0.24538 |
1.1874 |
Non-allergen |
Non-toxin |
100.00% |
0.004 |
46.5% (56.57%) |
| HLA-A*11:01 |
5.53 |
0.01 |
| HLA-A*30:01 |
38.3 |
0.04 |
| HLA-A*31:01 |
34.67 |
0.13 |
| HLA-A*68:01 |
153.42 |
0.72 |
| MASLVLARK |
633-641 |
1.00 |
A3 |
HLA-A*03:01 |
216.62 |
0.93 |
0.9 |
0.22 |
1.11 |
0.02816 |
1.3011 |
Non-allergen |
Non-toxin |
100.00% |
0.122 |
37.17% (44.94%) |
| HLA-A*11:01 |
37.49 |
0.46 |
| HLA-A*68:01 |
5.23 |
0.2 |
| ORF6 Protein |
| KVSIWNLDY |
23-31 |
0.30 |
A3 |
HLA-A*03:01 |
282.61 |
0.82 |
1.15 |
1.32 |
2.47 |
0.29343 |
0.8195 |
Non-allergen |
Non-toxin |
96.55% |
0.009 |
52.02% (56.63%) |
| HLA-A*11:01 |
189.72 |
0.91 |
| HLA-A*30:02 |
26.48 |
0.09 |
Note. CTL: Cytotoxic T-cells; IC50: Half-maximal inhibitory concentration; ORF: Open reading frame; Nsp: Nonstructural protein; MHC: Histocompatibility complex.
Table 3.
Predicted Top-Ranked HTL Epitopes
|
Epitope
|
Position
|
NetMHC II Percentile Rank
|
MHC Class II Allele
|
IC50 (nM)
|
TepiTool Percentile Rank
|
Antigenicity Score (Cut off Score, 0.4)
|
Allergenicity
|
Toxicity
|
IFNγ
|
IL-4
|
IL-10
|
Conservancy
|
Mean Entropy Score
|
Population Coverage
|
| S1 Domain |
| VVLSFELLHAPATVC |
499-513 |
0.08 |
HLA-DRB1*01:01 |
4.15 |
1.4 |
0.8618 |
Non-allergen |
Non-toxin |
Inducer (0.47729732) |
Inducer (0.28) |
Inducer (0.3505322) |
100.00% |
0.521 |
45.48% (20.88%) |
| HLA-DRB1*04:01 |
150.5 |
28 |
| HLA-DRB1*09:01 |
109.37 |
50 |
| HLA-DRB1*15:01 |
123.28 |
42 |
| HLA-DRB1*04:05 |
110.76 |
29 |
| HLA-DPA1*03:01/DPB1*04:02 |
143.5 |
35 |
| N Protein |
| QIAQFAPSASAFFGM |
303-317 |
3.050 |
HLA-DRB1*04:01 |
164.6 |
20 |
0.4032 |
Non-allergen |
Non-toxin |
Inducer (1.0) |
Inducer (0.31) |
Inducer (0.34858621) |
100.00% |
0.032 |
35.63% (16.55%) |
| HLA-DRB1*07:01 |
15.29 |
1.2 |
| HLA-DRB1*09:01 |
16.91 |
0.31 |
| HLA-DRB1*08:02 |
497.39 |
42 |
| HLA-DRB3*01:01 |
321.84 |
6.7 |
| HLA-DRB5*01:01 |
207.27 |
21 |
| HLA-DQA1*04:01/DQB1*04:02 |
70.02 |
43 |
| HLA-DQA1*05:01/DQB1*03:01 |
93.13 |
16 |
| NFKDQVILLNKHIDA |
345-359 |
3.00 |
HLA-DRB1*04:01 |
357.65 |
40 |
0.7693 |
Non-allergen |
Non-toxin |
Inducer (2.1796585) |
Inducer (0.25) |
Inducer (0.41974463) |
100.00% |
0.030 |
32.81% (22.66%) |
| HLA-DRB1*11:01 |
164.91 |
1.7 |
| HLA-DRB1*12:01 |
375.06 |
2.9 |
| HLA-DRB1*08:02 |
404.98 |
2.3 |
| HLA-DRB1*13:02 |
40 |
4.7 |
| Nsp4 Protein |
| AVGNICYTPSKLIEY |
128-142 |
0.12 |
HLA-DRB1*07:01 |
11.04 |
0.45 |
1.0892 |
Non-allergen |
Non-toxin |
Inducer (1.5539285) |
Inducer (1.51) |
Inducer (0.4329907) |
97.73% |
0.036 |
45.1% (26.15%) |
| HLA-DRB1*09:01 |
69.64 |
7.2 |
| HLA-DRB1*15:01 |
75.43 |
7.1 |
| HLA-DRB1*13:02 |
218 |
20 |
| KHFYWFFSNYLKRRV |
388-402 |
3.50 |
HLA-DRB1*07:01 |
21.21 |
5.8 |
0.4111 |
Non-allergen |
Non-toxin |
Inducer (1.0) |
Inducer (1.35) |
Inducer (1.1760984) |
100.00% |
0.0 |
47.22% (40.04%) |
| HLA-DRB1*11:01 |
71.71 |
20 |
| HLA-DRB1*15:01 |
15.01 |
3.8 |
| HLA-DRB1*08:02 |
230.22 |
22 |
| HLA-DRB1*04:05 |
35.82 |
11 |
| HLA-DRB3*02:02 |
133.58 |
18 |
| HLA-DRB5*01:01 |
16.47 |
1.4 |
| HLA-DPA1*01:03/DPB1*02:01 |
20.59 |
3.6 |
| HLA-DPA1*01:03/DPB1*04:01 |
38.01 |
- |
| HLA-DPA1*02:01/DPB1*01:01 |
49.83 |
5.7 |
| HLA-DPA1*02:01/DPB1*05:01 |
91.35 |
3.1 |
| HLA-DPA1*03:01/DPB1*04:02 |
36.89 |
5.4 |
| HLA-DQA1*01:01/DQB1*05:01 |
207.75 |
6.6 |
| Nsp12 Protein |
| SKGFFKEGSSVELKH |
425-439 |
1.80 |
HLA-DRB1*01:01 |
15.62 |
2.9 |
0.6212 |
Non-allergen |
Non-toxin |
Inducer (1.8951788) |
Inducer (0.29) |
Inducer (0.39050212) |
100.00% |
0.018 |
43.06% (24.85%) |
| HLA-DRB1*04:01 |
121.4 |
2 |
| HLA-DRB1*07:01 |
50.80 |
1.9 |
| HLA-DRB1*09:01 |
35.23 |
0.94 |
| NFKSVLYYQNNVFMS |
781-795 |
6.00 |
HLA-DRB1*01:01 |
42.82 |
25 |
0.6860 |
Non-allergen |
Non-toxin |
Inducer (2.001151) |
Inducer (0.25) |
Inducer (0.36805841) |
100.00% |
0.016 |
47.42% (24.48%) |
| HLA-DRB1*04:01 |
106.4 |
26 |
| HLA-DRB1*04:05 |
108.75 |
25 |
| HLA-DRB1*13:02 |
56.24 |
23 |
| HLA-DRB1*15:01 |
14.29 |
0.65 |
| HLA-DRB3*02:02 |
56.25 |
13 |
| HLA-DRB4*01:01 |
153.4 |
22 |
| HLA-DPA1*01:03/DPB1*04:01 |
87.46 |
- |
| HLA-DPA1*02:01/DPB1*02:01 |
195.64 |
- |
| HLA-DPA1*02:01/DPB1*05:01 |
271.65 |
- |
| HLA-DPA1*03:01/DPB1*04:02 |
92.21 |
20 |
| HLA-DQA1*01:01/DQB1*05:01 |
228.2 |
2.7 |
Note. MHC: Histocompatibility complex; HLA: Human leukocyte antigen; IC50: Half-maximal inhibitory concentration; IFNγ: Interferon gamma; IL: Interleukin; HTL: Helper T-cell; Nsp: Nonstructural protein.
Vaccine Formulation
The anti-SARS-CoV-2 MEV was scaffolded through linking the 11 CTLs, 7 HTLs, and 15 LBLs epitopes (Figure S1). The physicochemical properties of this construct revealed a strong antigenicity with no allergenicity (Table S1). Moreover, only one asparagine residue in position 432 (0.5508 score) was predicted asthe potential N-glycosylation site. Thus, the risk of epitope masking is extremely low. This residue belonged to one LBL and one conformational epitope of the ORF6 protein.
The query coverage scores between the vaccine construct and the human proteome and other species did not exceed more than 7% and 8%, respectively. As a favorable feature, a BLASTP score of < 37% is typically an indication of non-homology. Therefore, it decreases the induction of autoimmune responses in the host.
Chimeric Construct Modelling
The alpha helices, beta strands, and random coils accounted for 241 (42.21%), 80 (14.01%), and 250 (43.78%), respectively (Figure S2), showing the construct’s ability to form various intrachain and interchain secondary structures.
Following 3D modelling, the confidence score (the C-score) of the five top-ranked models was estimated in the range of -1.20 to -3.39, where a higher value indicates a higher confidence. Accordingly, model 3 (C-score: -1.20) was subjected to refinement analysis (Figure S3).
The final refined model had the quality scores 0.9488, 2.507, 0.417, 1.4, 26.3, and 92.3 for the high-accuracy global distance test, MolProbity, RMSD, poor rotamers, clash score, and Rama favored, respectively. A Ramachandran plot analysis located 89.8% of residues in energetically favored regions, 8.8% in allowed regions, and only 1.4% in disallowed regions. The quality assessment of this model estimated a Z-score of -3.59 using ProSA analysis, suggesting that the structure lies in the range of experimentally validated protein models. An overall quality factor of 86.535 was predicted by the ERRAT program as well (Figure S3).
Conformational Epitope Prediction
The five conformational epitopes were identified with a surface-exposed position (protrusion scores in the range of 0.544–0.78) (Table S2 and Figure S4), indicating their potential to induce an immune response through recognition by antibodies.
Protein-Protein Docking
The MEV interaction with the key immune receptors was evaluated by docking analysis (Table 4). The most favorable interactions were observed for TLR4 (Figure S5 and Table S3), TLR8, and HLA-A*03:01 (Figures S6 and S7 and Tables S4 and S5). The results were confirmed by a low calculated RMSD (i.e., 0.134 Å). The RMSD values ≤ 2 Å were considered a validation of the docking protocol.
Table 4.
A Comprehensive Overview of Molecular Docking Results
Docked Complex of MEV-TLR
s:
|
Energy of Docking (kcal/mol)
|
Dissociation Constant (Kd)
|
Binding Energy (ΔG in kcal/mol)
|
Contacts (VDW Pairs in≥-0.40Å)
|
H Bonds
|
Salt Bridges
|
Docked Complex of MEV -MHC I/II:
|
Energy of Docking (kcal/mol)
|
Dissociation Constant (Kd)
|
Binding Energy (ΔG in kcal/mol)
|
Contacts (VDW Pairs in≥-0.40Å)
|
H Bonds
|
Salt Bridges
|
| TLR2 |
-810.74 |
3.2e-57 |
-80.1 |
10335 |
26 |
1 |
HLA-A*03:01 |
-1482.33 |
5.9e-18 |
-24.4 |
2334 |
20 |
3 |
| TLR3 |
-1042.85 |
1.2e-19 |
-26.8 |
3011 |
8 |
0 |
HLA-A*11:01 |
-1644.99 |
1.8e-10 |
-13.8 |
954 |
7 |
1 |
| TLR4 |
-1419.94 |
1.8e-59 |
-83.3 |
12925 |
25 |
2 |
HLA-A*68:01 |
-1358.14 |
7.8e-09 |
-11.5 |
588 |
4 |
- |
| TLR5 |
-1307.63 |
1.5e-41 |
-57.9 |
7535 |
20 |
- |
HLA-B*35:01 |
-857.14 |
3.7e-12 |
-16.2 |
996 |
4 |
1 |
| TLR7 |
-277.09 |
2.5e-47 |
-66.1 |
6994 |
20 |
1 |
HLA-DRB1*01:01 |
-1984.55 |
5.6e-10 |
-13.1 |
558 |
4 |
- |
| TLR8 |
-968.62 |
1.6e-56 |
-79.1 |
14076 |
30 |
1 |
HLA-DRB1*09:01 |
-2468.18 |
9.6e-12 |
-15.6 |
811 |
6 |
1 |
| ACE2 |
-1995.73 |
8.5e-30 |
-41.2 |
5402 |
94 |
9 |
|
Note. TLR: Toll-like receptor; MEV: Epitope vaccine construct; MHC: Histocompatibility complex; ACE: Angiotensin-converting enzyme 2; HLA: Human leukocyte antigen.
In total, these results highlight the construct’s ability to activate the innate immunity through plasma membrane or endosome-related TLRs. It is in alignment with its immunodominant epitopes, especially from the S1 (as a surface viral protein) and N (wrapped around the viral genome) proteins. The MEV also showed good interactions with MHC I and II, particularly HLA-A*03:01, HLA-DRB1*09:01, and HLA-DRB1*01:01. The potent MEV-MHC I/II interactions suggest an effective CD8+ and CD4+ T cell activation following the vaccination to enhance the immune responses. More details are shown in Figures S8-S16 and Tables S6-S14.
The CABS-flex analysis indicated different flexibility and root-mean square fluctuation values upon vaccine construct-immune receptors. Importantly, the key amino acid residues demonstrated a relatively low root-mean square fluctuation in all complexes as stable and rigid interactions (Figure S17 and Table S15).
On the other hand, the iMODS provided more information about the dynamic behavior of each complex (Table S16 and Figures S18-S29). Accordingly, the most stable complexes were HLA-A*68:01, HLA-A*03:01, HLA-DRB1*01:01, HLA-B*35:01, and TLR2-7, with relatively low peaks in the deformability graph (~0.8 Å deformability index). The B-factor plot analysis also verified these results.
In addition, the HLA-A*11:01, HLA-A*03:01, HLA-B*35:01, HLA-A*68:01, and HLA-DRB1*09:01 complexes were predicted as the most stable complexes by the eigenvalue and variance plots. These parameters are inversely related, with a lower eigenvalue indicating less required energy for deformation and a higher variance suggesting greater flexibility.
A high correlation between the amino acid residues is typically indicated by more red areas in the covariance matrix graph. Thus, all docked complexes showed stable correlations. Moreover, darker gray dots represent stronger connections in the elastic network model. This stiffness was maintained in all complexes, pointing to stable interactions as a critical factor for downstream immune activation.
Additionally, the favorable interactions were observed between MEV and ACE2. Interestingly, the LBL epitopes of S1 (i.e., AAYYVGYL, KGIYQTSNFRVQPTES, and NDLCFTNVYADSFVIR) made considerable interactions with the ACE2 (Table S17). The superiority of these results relative to ACE2 interactions with the spike of SARS-CoV-2 wild-type and multiple variants (19) can imply the promising potential of this vaccine candidate to prevent virus-cell interactions.
Multi-Epitope Vaccine Construct Single-Chain Variable Fragment Interaction Simulation
The multiple paratope-epitope interactions were predicted in the MEV (as an antigen) and scFv (as the antibody) as 12 hydrogen bonds, 2 salt bridges, and 340 contacts (VDW pairs in ≥ 0.40 Å; Figure S30). The thermodynamic statistics of this interaction included the energy of docking (kcal/mol) of -284.25, dissociation constant (Kd) equal to 7.1e-09, and binding energy (ΔG) equal to -11.6 kcal/mol.
Herein, THR52 and LYS95 residues participate in the interaction as the complementary determining regions 2 and 3 from the variable domain of the scFv’s heavy chain (VH), respectively (Figure S30). Yaghoobizadeh et al (19) also verified the VH importance to interact with SARS-CoV-2 RBD. More interestingly, this anti-RBD scFv made the four H bonds with one of the predicted immunodominant S1 epitopes (i.e., VVLSFELLHAPATVC). This epitope was also classified as an efficient MHC II-binder with good immunogenicity scores (Table 3). The molecular dynamicssimulation confirmed the binding mode and the conformational flexibility of this interaction as well (Figure S31).
In Silico Cloning and Messenger Ribonucleic Acid Stability
The in silico cloning of this ~1.73 kbp construct was performed, resulting in a ~6.99 kbp clone (Figure S32). The codon optimization changed the GC% and CAI from 49.10% to 49.21% and from 1.0 to 0.93, respectively. The effective translation and expression of the MEV could be ensured with regard to the ideal range of 30%–70% and 0.8–1.0 for GC% and CAI, respectively.
Thereafter, the mRNA secondary structure was predicted with a single-stranded value and the minimum-free energy equal to 17.20 ± 15.09 and -529.25 kcal/mol, respectively (Figure S33). This structure contained multiple hairpins, loops, and stem-loops. However, not only were no hairpins or pseudoknots predicted at its 5’ end (Figure S33b), but also a high possibility of single-stranded formation was confirmed at this end (Figure S33c). Both features lead to easier ribosome accessibility to the start codon and initiation site of translation for mRNA. Additionally, the proximity of both ends through the formation of multiple bonds enhances the translation rates by stabilizing the initiation complex. This base-pairing was also confirmed by the energy dot plot (Figure S33e, the upper right triangle) and the circular energy diagram (Figure S33d).
Immunogenic Behavior Profiling
The immune response simulation revealed a strong reaction following a booster vaccination (Figure S34). As expected, the titer of various immunoglobulins was enhanced, especially following the 3rd and 4th boosters. Moreover, not only was a reasonable humoral immunity observed by increased IgM and IgG, but also immunocomplexes indicated a good performance in antigen clearance. These findings suggest an increase in antibody specificity and affinity maturation toward antigens. The enhancement in various B-cell populations, their performance, and isotype switching also supports this observation.
The TC/TH populations showed the positive changes as well. The TH population has a critical role in boosting the memory and stimulating the adaptive immunity arms, thus promoting the B-cell proliferation and antibody class switching.
The results also highlighted the construct efficiency in activating innate immune cells, that is, antigen-presenting cells (APCs), and natural killer cells. It enhances the subsequent immune responses against the immunogen and controls the associated infectious agent.
Finally, the vaccine induced a robust cytokine response, especially by increasing IFNγ levels. It aligned with the IFNγ-inducing ability of selected epitopes. The cytokine induction reflects a favorable immune response, likely resulting from TLR-MEV interactions (13). Furthermore, a negligible Simpson index (D) score was predicted, alleviating the risk of excessive cytokine production and the following immune complications (4).
Discussion
The introduction of the smallpox vaccine by Edward Jenner’s innovation in the 18th century laid the foundations for the golden age of vaccine production. Moreover, the computational approaches further revolutionized vaccine design and drug discovery and decreased the timeline for potent intervention development (5,6).
Therefore, following the global health crisis caused by SARS-CoV-2 as a Public Health Emergency of International Concern, enormous antiviral drugs have been designed (7,11,29,41) or repurposed (42), and various therapeutic antibodies have been introduced (10,12,19) and used in commercial (https://www.antibodies-online.com/areas/infectious-disease/covid-19/sars-cov-2-antibodies/). On the other hand, over 50 vaccines and 242 candidates have been introduced, employing diverse production platforms (https://covid19.trackvaccines.org/). Notably, protein subunit vaccines, such as COVOVAX, account for 36% of the approved vaccines, making them a main contributor to pooling anti-SARS-CoV-2 vaccines (4,11,13,18,27,30).
On the other hand, epitope-based vaccines have attracted significant attention to enhance immunization efficiency via combining immunodominant and conserved epitopes. This second-generation production platform enables a faster, cost-effective, more targeted, and robust response to viral diversity. In addition, it minimizes the side effects, especially for immunocompromised individuals. The inclusion of suitable adjuvants boosts the vaccine immunogenicity as well (4,5,6,13,23,29). Accordingly, epitope-based vaccines have been successfully applied against various parasites (26,33,37), bacteria (24,38), viruses (25,40), allergic conditions (43,44), and cancers (45,46).
Many studies have also reported the development of MEV against SARS-CoV-2. However, there is a high variation in a potent formulation design using the structural (5,13,27,29,36), nonstructural (39), or a combination of structural, nonstructural, or accessory (4,30,32) proteins. This study also introduced a novel MEV formulation using the immunodominant epitopes of SARS-CoV-2 proteins. To our knowledge, no MEV incorporating epitopes from these proteins has been reported with a potential capacity as pan-species prophylactics.
The promising physicochemical properties are critical for vaccine design and support vaccine efficiency. These properties affect the efficiency, safety, antigen processing, and presentation. The inclusion of 92 positively charged (cationic) groups, compared to 31 negatively charged (anionic) groups, enables the construct to make strong electrostatic interactions with APCs. The hydrophilic nature of the construct (GRAVY index -0.478) also indicates a good interaction with solvents (e.g., water and blood) to facilitate its delivery to target sites. Moreover, an appropriate aliphatic index (65.46) suggests its stability for in vitro studies. An instability index (II) < 40 (34.80) ensures an adequate stability for immune stimulation as well (5,13,27).
The molecular weight lower than 110 kDa mainly makes the recombinant constructs suitable candidates. Hence, a molecular weight of ~65 kDa is considered a strength point of the designed MEV (4,13,31,32,36).
The recognition of pathogen-associated molecular patterns via pattern recognition receptors is necessary for APC activation and the following immune responses. Vaccine constructs containing pathogen-associated molecular patterns could also activate these pathways to enhance antigen presentation to the effector cells (5,27,36,47,48).
Among pattern recognition receptors, TLRs are important innate immunity mediators, bridging adaptive immune responses. The human genome encodes 10 functional TLRs, with six TLRs (1, 2, 4, 5, 6, and 10) placed on the cell membrane, and the expression of the remaining TLRs (i.e., 3, 7, 8, and 9) occurs in intracellular biocompartments, such as endosomes. Many of the innate immunity cells, non-hematopoietic cells, and B-cells and T-cells are equipped with these receptors to recognize a vast range of molecular patterns, including viral nucleic acids (TLR3, 7, 8, 9) and structural/non-structural proteins (TLR2, 4). The TLR activation will induce proinflammatory/antiviral cytokines and chemokine production via signal transduction. Accordingly, adaptive immunity cells will be functionalized (47-49). Similar to the current study, the interactions between anti-SARS-CoV-2 vaccine constructs and these TLRs have been extensively investigated previously (4,13,27,30). Similarly, one study also evaluated the anti-SARS-CoV-2 vaccine-TLR5 interaction (36).
Conclusion
The SARS-CoV-2 pandemic has driven the development of diverse prophylactics and therapeutics, including those via in silico approaches. In this regard, this study presented a novel MEV formulation showing good efficiency by multiple computational and immunoinformatic analyses. The strong in silico interactions were also observed with a validated scFv as a preliminary computational indication of the possible in vivo efficiency. On the other hand, in silico mutagenesis in non-critical amino acid residues could improve vaccine properties. Meanwhile, if its potency is confirmed in wet-lab experiments, it might be considered a promising anti-SARS-CoV-2 formulation with a pan-species activity.
Competing Interests
All authors declare that they have no conflict of interests.
Funding
This work was supported by the Center for International Scientific Studies and Collaboration, Ministry of Science, Research and Technology.
Supplementary Files
Supplementary file 1 contains Figures S1-S34 and Tables S1-S17
(pdf)
References
- Fang R, Yang X, Guo Y, Peng B, Dong R, Li S. SARS-CoV-2 infection in animals: patterns, transmission routes, and drivers. Eco Environ Health 2024; 3(1):45-54. doi: 10.1016/j.eehl.2023.09.004 [Crossref] [ Google Scholar]
- V’Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol 2021; 19(3):155-70. doi: 10.1038/s41579-020-00468-6 [Crossref] [ Google Scholar]
- Yaghoobizadeh F, Ardakani MR, Ranjbar MM, Galehdari H, Khosravi M. Expression, purification, and study on the efficiency of a new potent recombinant scFv antibody against the SARS-CoV-2 spike RBD in E coli BL21. Protein Expr Purif 2023; 203:106210. doi: 10.1016/j.pep.2022.106210 [Crossref] [ Google Scholar]
- Almofti YA, Abd-Elrahman KA, Eltilib EE. Vaccinomic approach for novel multi epitopes vaccine against severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). BMC Immunol 2021; 22(1):22. doi: 10.1186/s12865-021-00412-0 [Crossref] [ Google Scholar]
- Al Zamane S, Nobel FA, Jebin RA, Amin MB, Somadder PD, Antora NJ. Development of an in silico multi-epitope vaccine against SARS-COV-2 by précised immune-informatics approaches. Inform Med Unlocked 2021; 27:100781. doi: 10.1016/j.imu.2021.100781 [Crossref] [ Google Scholar]
- Kar T, Narsaria U, Basak S, Deb D, Castiglione F, Mueller DM. A candidate multi-epitope vaccine against SARS-CoV-2. Sci Rep 2020; 10(1):10895. doi: 10.1038/s41598-020-67749-1 [Crossref] [ Google Scholar]
- Pedrini M, Pozzi L, Sacchi F, Citarella A, Fasano V, Seneci P. Design, synthesis and in vitro validation of bivalent binders of SARS-CoV-2 spike protein: obeticholic, betulinic and glycyrrhetinic acids as building blocks. Bioorg Med Chem 2025; 121:118124. doi: 10.1016/j.bmc.2025.118124 [Crossref] [ Google Scholar]
- Ysrafil Y, Imran AK, Wicita PS, Kamba V, Mohamad F, Ismail I. Mosaic vaccine design targeting mutational spike protein of SAR-SCoV-2: an immunoinformatics approach. Bioimpacts 2025; 15:26443. doi: 10.34172/bi.2023.26443 [Crossref] [ Google Scholar]
- Amini Pouya M, Afshani SM, Salek Maghsoudi A, Hassani S, Mirnia K. Classification of the present pharmaceutical agents based on the possible effective mechanism on the COVID-19 infection. Daru 2020; 28(2):745-64. doi: 10.1007/s40199-020-00359-4 [Crossref] [ Google Scholar]
- Feng B, Li C, Zhang Z, Huang Y, Liu B, Zhang Z. A shark-derived broadly neutralizing nanobody targeting a highly conserved epitope on the S2 domain of sarbecoviruses. J Nanobiotechnology 2025; 23(1):110. doi: 10.1186/s12951-025-03150-2 [Crossref] [ Google Scholar]
- Kirar M, Singh H, Sehrawat N. Virtual screening and molecular dynamics simulation study of plant protease inhibitors against SARS-CoV-2 envelope protein. Inform Med Unlocked 2022; 30:100909. doi: 10.1016/j.imu.2022.100909 [Crossref] [ Google Scholar]
- Pitsillou E, El-Osta A, Hung A, Karagiannis TC. Epimaps of the SARS-CoV-2 receptor-binding domain mutational landscape: insights into protein stability, epitope prediction, and antibody binding. Biomolecules 2025; 15(2):301. doi: 10.3390/biom15020301 [Crossref] [ Google Scholar]
- Sarvmeili J, Baghban Kohnehrouz B, Gholizadeh A, Shanehbandi D, Ofoghi H. Immunoinformatics design of a structural proteins driven multi-epitope candidate vaccine against different SARS-CoV-2 variants based on fynomer. Sci Rep 2024; 14(1):10297. doi: 10.1038/s41598-024-61025-2 [Crossref] [ Google Scholar]
- Soares SR, da Silva Torres MK, Lima SS, de Sarges KM, Dos Santos EF, de Brito M. Antibody response to the SARS-CoV-2 spike and nucleocapsid proteins in patients with different COVID-19 clinical profiles. Viruses 2023; 15(4):898. doi: 10.3390/v15040898 [Crossref] [ Google Scholar]
- Gorkhali R, Koirala P, Rijal S, Mainali A, Baral A, Bhattarai HK. Structure and function of major SARS-CoV-2 and SARS-CoV proteins. Bioinform Biol Insights 2021; 15:11779322211025876. doi: 10.1177/11779322211025876 [Crossref] [ Google Scholar]
- Català M, Mercadé-Besora N, Kolde R, Trinh NT, Roel E, Burn E. The effectiveness of COVID-19 vaccines to prevent long COVID symptoms: staggered cohort study of data from the UK, Spain, and Estonia. Lancet Respir Med 2024; 12(3):225-36. doi: 10.1016/s2213-2600(23)00414-9 [Crossref] [ Google Scholar]
- Ceban F, Kulzhabayeva D, Rodrigues NB, Di Vincenzo JD, Gill H, Subramaniapillai M. COVID-19 vaccination for the prevention and treatment of long COVID: a systematic review and meta-analysis. Brain Behav Immun 2023; 111:211-29. doi: 10.1016/j.bbi.2023.03.022 [Crossref] [ Google Scholar]
- Nag A, Varun P. Designing potential long-term vaccine targets against emerging variants of SARS-CoV-2 using an immunoinformatics approach. Next Res 2025; 2(1):100112. doi: 10.1016/j.nexres.2024.100112 [Crossref] [ Google Scholar]
- Yaghoobizadeh F, Roayaei Ardakani M, Ranjbar MM, Khosravi M, Galehdari H. Development of a potent recombinant scFv antibody against the SARS-CoV-2 by in-depth bioinformatics study: paving the way for vaccine/diagnostics development. Comput Biol Med 2024; 170:108091. doi: 10.1016/j.compbiomed.2024.108091 [Crossref] [ Google Scholar]
- Zhao F, Zhang Y, Zhang Z, Chen Z, Wang X, Wang S. Epitope-focused vaccine immunogens design using tailored horseshoe-shaped scaffold. J Nanobiotechnology 2025; 23(1):119. doi: 10.1186/s12951-025-03200-9 [Crossref] [ Google Scholar]
- Rahman MS, Hoque MN, Islam MR, Akter S, Rubayet Ul Alam AS, Siddique MA. Epitope-based chimeric peptide vaccine design against S, M and E proteins of SARS-CoV-2, the etiologic agent of COVID-19 pandemic: an in-silico approach. PeerJ 2020; 8:e9572. doi: 10.7717/peerj.9572 [Crossref] [ Google Scholar]
- Williams JM, Chen YJ, Cho WJ, Tai AW, Tsai B. Reticulons promote formation of ER-derived double-membrane vesicles that facilitate SARS-CoV-2 replication. J Cell Biol 2023; 222(7):e202203060. doi: 10.1083/jcb.202203060 [Crossref] [ Google Scholar]
- Sun Q, Huang Z, Yang S, Li Y, Ma Y, Yang F. Bioinformatics-based SARS-CoV-2 epitopes design and the impact of spike protein mutants on epitope humoral immunities. Immunobiology 2022; 227(6):152287. doi: 10.1016/j.imbio.2022.152287 [Crossref] [ Google Scholar]
- Touhidinia M, Sefid F, Bidakhavidi M. Design of a Multi-epitope vaccine against Acinetobacter baumannii using immunoinformatics approach. Int J Pept Res Ther 2021; 27(4):2417-37. doi: 10.1007/s10989-021-10262-4 [Crossref] [ Google Scholar]
- Yu C, Wu Q, Xin J, Yu Q, Ma Z, Xue M. Designing a smallpox B-cell and T-cell multi-epitope subunit vaccine using a comprehensive immunoinformatics approach. Microbiol Spectr 2024; 12(6):e0046524. doi: 10.1128/spectrum.00465-24 [Crossref] [ Google Scholar]
- Danazumi AU, Iliyasu Gital S, Idris S, Bs Dibba L, Balogun EO, Górna MW. Immunoinformatic design of a putative multi-epitope vaccine candidate against Trypanosoma brucei gambiense. Comput Struct Biotechnol J 2022; 20:5574-85. doi: 10.1016/j.csbj.2022.10.002 [Crossref] [ Google Scholar]
- Tourani M, Samavarchi Tehrani S, Movahedpour A, Rezaei Arablouydareh S, Maleksabet A, Savardashtaki A. Design and evaluation of a multi-epitope vaccine for COVID-19: an in-silico approach. Health Sci Monit 2023; 2(3):180-204. doi: 10.61186/hsm.2.3.180 [Crossref] [ Google Scholar]
- Corripio-Miyar Y, Hayward A, Lemon H, Sweeny AR, Bal X, Kenyon F. Functionally distinct T-helper cell phenotypes predict resistance to different types of parasites in a wild mammal. Sci Rep 2022; 12(1):3197. doi: 10.1038/s41598-022-07149-9 [Crossref] [ Google Scholar]
- Adelusi TI, Ogunlana AT, Oyewole MP, Ojo TO, Olaoba OT, Oladipo EK. Designing of an innovative conserved multiepitope subunit vaccine targeting SARS-CoV-2 glycoprotein and nucleoprotein through immunoinformatic. Sci Rep 2025; 15(1):2563. doi: 10.1038/s41598-024-72495-9 [Crossref] [ Google Scholar]
- Oso BJ, Olaoye IF, Ogidi CO. In silico design of a vaccine candidate for SAR S-CoV-2 based on multiple T-cell and B-cell epitopes. Arch Razi Inst 2021; 76(5):1191-202. doi: 10.22092/ari.2020.351605.1526 [Crossref] [ Google Scholar]
- Pahlavan Y, Yeganeh O, Asghariazar V, Karami C. Multi-epitope vaccine against SARS-CoV-2 targeting the spike RBD: an immunoinformatics approach. Future Sci OA 2024; 10(1):FSO939. doi: 10.2144/fsoa-2023-0081 [Crossref] [ Google Scholar]
- Palatnik-de-Sousa I, Wallace ZS, Cavalcante SC, Ribeiro MP, Silva J, Cavalcante RC. A novel vaccine based on SARS-CoV-2 CD4 + and CD8 + T cell conserved epitopes from variants Alpha to Omicron. Sci Rep 2022; 12(1):16731. doi: 10.1038/s41598-022-21207-2 [Crossref] [ Google Scholar]
- Shams M, Nourmohammadi H, Asghari A, Basati G, Majidiani H, Naserifar R. Construction of a multi-epitope protein for human Toxocaracanis detection: immunoinformatics approach multi-epitope construct for T canis serodiagnosis. Inform Med Unlocked 2021; 26:100732. doi: 10.1016/j.imu.2021.100732 [Crossref] [ Google Scholar]
- Imberty A, Hardman KD, Carver JP, Pérez S. Molecular modelling of protein-carbohydrate interactions Docking of monosaccharides in the binding site of concanavalin A. Glycobiology 1991; 1(6):631-42. doi: 10.1093/glycob/1.6.631 [Crossref] [ Google Scholar]
- Santra D, Maiti S. Molecular dynamic simulation suggests stronger interaction of Omicron-spike with ACE2 than wild but weaker than Delta SARS-CoV-2 can be blocked by engineered S1-RBD fraction. Struct Chem 2022; 33(5):1755-69. doi: 10.1007/s11224-022-02022-x [Crossref] [ Google Scholar]
- Yazdani Z, Rafiei A, Yazdani M, Valadan R. Design an efficient multi-epitope peptide vaccine candidate against SARS-CoV-2: an in-silico analysis. Infect Drug Resist 2020; 13:3007-22. doi: 10.2147/idr.S264573 [Crossref] [ Google Scholar]
- Gorai S, Das NC, Gupta PS, Panda SK, Rana MK, Mukherjee S. Designing efficient multi-epitope peptide-based vaccine by targeting the antioxidant thioredoxin of bancroftian filarial parasite. Infect Genet Evol 2022; 98:105237. doi: 10.1016/j.meegid.2022.105237 [Crossref] [ Google Scholar]
- Mandal M, Mandal S. MM-GBSA and QM/MM simulation-based in silico approaches for the inhibition of Acinetobacter baumannii class D OXA-24 β-lactamase using antimicrobial peptides melittin and RP-1. Chem Phys Impact 2024; 8:100401. doi: 10.1016/j.chphi.2023.100401 [Crossref] [ Google Scholar]
- Qazi S, Das S, Khuntia BK, Sharma V, Sharma S, Sharma G. In silico molecular docking and molecular dynamic simulation analysis of phytochemicals from Indian foods as potential inhibitors of SARS-CoV-2 RdRp and 3CLpro. Nat Prod Commun 2021; 16(9):1934578X211031707. doi: 10.1177/1934578X211031707 [Crossref] [ Google Scholar]
- Ryan N, Pratiwi SE, Mardhia M, Ysrafil Y, Liana DF, Mahyarudin M. Immunoinformatics approach for design novel multi-epitope prophylactic and therapeutic vaccine based on capsid proteins L1 and L2 and oncoproteins E6 and E7 of human papillomavirus 16 and human papillomavirus 18 against cervical cancer. Osong Public Health Res Perspect 2024; 15(4):307-28. doi: 10.24171/j.phrp.2024.0013 [Crossref] [ Google Scholar]
- Moschovou K, Antoniou M, Chontzopoulou E, Papavasileiou KD, Melagraki G, Afantitis A. Exploring the binding effects of natural products and antihypertensive drugs on SARS-CoV-2: an in-silico investigation of main protease and spike protein. Int J Mol Sci 2023; 24(21):15894. doi: 10.3390/ijms242115894 [Crossref] [ Google Scholar]
- Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J Chem Inf Model 2020; 60(6):3277-86. doi: 10.1021/acs.jcim.0c00179 [Crossref] [ Google Scholar]
- Qin QZ, Tang J, Wang CY, Xu ZQ, Tian M. Construction by artificial intelligence and immunovalidation of hypoallergenic mite allergen Der f 36 vaccine. Front Immunol 2024; 15:1325998. doi: 10.3389/fimmu.2024.1325998 [Crossref] [ Google Scholar]
- Sircar G, Ghosh N, Saha S. Designing next-generation vaccines against common pan-allergens using in silico approaches. Monoclon Antib Immunodiagn Immunother 2022; 41(5):231-42. doi: 10.1089/mab.2021.0033 [Crossref] [ Google Scholar]
- Heidary F, Tourani M, Hejazi-Amiri F, Khatami SH, Jamali N, Taheri-Anganeh M. Design of a new multi-epitope peptide vaccine for non-small cell Lung cancer via vaccinology methods: an in-silico study. Mol Biol Res Commun 2022; 11(1):55-66. doi: 10.22099/mbrc.2022.42468.1697 [Crossref] [ Google Scholar]
- Sanami S, Azadegan-Dehkordi F, Rafieian-Kopaei M, Salehi M, Ghasemi-Dehnoo M, Mahooti M. Design of a multi-epitope vaccine against cervical cancer using immunoinformatics approaches. Sci Rep 2021; 11(1):12397. doi: 10.1038/s41598-021-91997-4 [Crossref] [ Google Scholar]
- Kircheis R, Planz O. The role of toll-like receptors (TLRs) and their related signaling pathways in viral infection and inflammation. Int J Mol Sci 2023; 24(7):6701. doi: 10.3390/ijms24076701 [Crossref] [ Google Scholar]
- Sameer AS, Nissar S. Toll-like receptors (TLRs): structure, functions, signaling, and role of their polymorphisms in colorectal cancer susceptibility. Biomed Res Int 2021; 2021:1157023. doi: 10.1155/2021/1157023 [Crossref] [ Google Scholar]
- Duan T, Du Y, Xing C, Wang HY, Wang RF. Toll-like receptor signaling and its role in cell-mediated immunity. Front Immunol 2022; 13:812774. doi: 10.3389/fimmu.2022.812774 [Crossref] [ Google Scholar]