Avicenna Journal of Clinical Microbiology and Infection. 10(2):49-57.
doi: 10.34172/ajcmi.3440
Original Article
Genome Investigation of Brucella melitensis Strains Isolated From Brucellosis Patients in Kuwait
Abu Salim Mustafa 1, *  , Mohd Wasif Khan 2, Nazima Habibi 3
, Mohd Wasif Khan 2, Nazima Habibi 3
Author information:
1Department of Microbiology, Faculty of Medicine, Health Sciences Centre, Kuwait University, Jabriya, Kuwait
2OMICS Research Unit and Research Core Facility, Faculty of Medicine, Health Sciences Centre, Kuwait University, Jabriya, Kuwait
3Biotechnology Program, Environment and Life Science Research Centre, Kuwait Institute for Scientific Research, Shuwaikh, Kuwait
Abstract
Background: Human brucellosis is present on all inhabited continents with high prevalence in many areas of the world, including Kuwait and the Middle East. To implement proper control measures, the identification and characterization of Brucella species and genotypes are required through a reliable and rapid subtyping method. In previous studies, whole-genome sequencing (WGS) has shown its potential as an epidemiological typing tool. Using WGS data, this study aimed to identify the species, phage sequences, putative antibiotic resistance genes, virulence factors, and genotypes of Brucella melitensis strains isolated from patients in Kuwait and other countries.
Methods: Five B. melitensis isolates of Kuwaiti origin and 31 other isolates of B. melitensis originating from 28 countries were analyzed using whole genome-based approaches for genotypic identification and typing. In-silico techniques were used to identify the sequences for phages, antibiotic resistance genes, virulence factors, and genotypes using multilocus sequence typing (MLST) and whole-genome single-nucleotide polymorphism (wgSNP).
Results: The analysis of WGS data demonstrated that all five Kuwaiti isolates belonged to the non-vaccine strains of B. melitensis. Furthermore, the data represented the presence of two phage sequences, two antibiotic resistance genes, and 51 virulence factors in Kuwaiti isolates. Eventually, the genotypes of all isolates were identified based on MLST and wgSNP analysis, and wgSNP analysis suggested the possible areas/countries of origin of Kuwaiti isolates.
Conclusion: WGS data can be used to characterize Brucella isolates, and molecular techniques can be applied in-silico to rapidly identify and classify Brucella into species and genotypes and trace the possible origin of the isolates.
Keywords: Brucella melitensis, Whole genome sequencing, Genotypes
Copyright and License Information
© 2023 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Please cite this article as follows: Mustafa AS, Khan MW, Habibi N. Genome investigation of Brucella melitensis strains isolated from brucellosis patients in Kuwait. Avicenna J Clin Microbiol Infect. 2023; 10(2):49-57. doi:10.34172/ajcmi.2023.3440
Introduction
Brucellosis is a chronic infectious disease caused by Brucella spp., a Gram-negative facultative intracellular pathogenic bacterium (1). Globally, about 500 000 incident cases of human brucellosis are reported per year, but the true incidence is estimated to be 5 000 000 to 12 500 000 cases annually (2). This infectious disease is present on all inhabited continents with high prevalence in many areas of the world (3).The areas at high risk of brucellosis infection are the countries of the Middle East, the Mediterranean region, Central and South America, Central and South East Asia, Sub-Saharan Africa, and the Caribbean (3).
Human brucellosis is commonly caused by three genetically and phenotypically closely related Brucella species [i.e. B. melitensis, B. abortus, and B. suis] (2), but B. melitensis is most pathogenic to humans and has been the main cause of disease in many countries (4,5). Infected domestic animals, primarily cattle, sheep, goats, and camels, as well as wild animals, are the natural reservoir of Brucella (1,2). The brucellosis infection is mainly caused by contact with infected animals or through the consumption of contaminated dairy products (2).
Species identification and genotyping of Brucella isolates are important for epidemiologic surveillance in Brucella endemic regions (6,7) Although biochemical assays have identified three biovars of B. melitensis, the limited genetic diversity and high DNA/DNA homology among Brucella genomes have made its genotyping a challenging task (8). Over the years, several approaches such as multilocus variable-number tandem repeat analysis (MLVA) and multilocus sequence typing (MLST) have been used to determine the genetic diversity among different species and the strains of this bacteria (9,10). In recent years, whole-genome sequencing (WGS) has emerged as a novel tool for the complete genetic characterization of an organism (11). It has the benefit of capturing nearly all the genetic variations and identifying the maximum amount of potentially informative loci (12). Hence, WGS provides a better resolution at the subspecies level (13). A promising approach that is capable of being incorporated into high-throughput assays is the use of single-nucleotide polymorphisms (SNPs). In previous studies, it was shown that WGS typing can achieve greater performance in terms of speed and accuracy than other traditional methods (12), and it was utilized to determine the phylogeny of B. melitensis across the globe (12).
Brucellosis is among the commonly reported infectious diseases in Kuwait and other neighboring countries of the Middle East (3,14). The population of Kuwait is highly diverse with about 30% Kuwaiti nationals and 70% expatriates (https://www.paci.gov.kw/stat). Among the expatriates, 40.4%, 27.2%, and 2% are Asians, Arabs, and others, respectively (https://www.paci.gov.kw/stat), suggesting many possible routes for the entry of Brucella in Kuwait. In this study, it was attempted to identify those routes of Brucella entry by finding the genotypes involved in Brucellosis in Kuwait. This would be important in endemic surveillance and control measures. Furthermore, we have compared our B. melitensis strains with other strains available at the National Center for Biotechnology Information (NCBI) genome database based on whole-genome SNPs (wgSNPs) and phylogenetic analysis. Moreover, it was also aimed to find out other genomic features such as genes specific in vaccine strains, phage sequences, putative antibiotic resistance genes, and traditional MLST profiling.
Materials and Methods
Strains
Five strains were selected from our previous study (9) to determine the genotypes of B. melitensis strains isolated from brucellosis patients in Kuwait. The assembly statistics of these strains have been described previously (14). The nucleotide FASTA, amino acid FASTA, and raw FASTQ files, corresponding to these Kuwaiti strains, were downloaded from the NCBI repository (15,16). A nucleotide FASTA file of 31 other strains of B. melitensis (originating from 28 countries) and one B. abortus strain, an outlier as shown by Georgi et al (17), were downloaded from NCBI genomes (16). The criteria for selecting B. melitensis isolates were 13 strains belonging to five genotypes of different geographical origins (12), one strain from each of the 17 countries (17), and one strain of Lebanon origin (18). For three strains (BwIM_DZA_50, BwIM_ROU_18, and BwIM_IND_58) included in the study of Georgi et al (17), the genome assemblies were not present in the NCBI database; hence, the raw FASTQ files were downloaded from the Sequence Read Archive of NCBI.
Brucella melitensis Specific Genetic Marker Identification
Brucella melitensis-specific IS711 sequence and the product of the primer pair UF1 and UR1 which can differentiate between B. melitensis and B. abortus (19) were extracted using the primers from the 16M strain (Table 1). The raw reads of Kuwaiti strains were mapped to these sequences using the k-mer alignment (KMA) program to confirm the Brucella species (20). To differentiate these strains from the vaccine strain, the rpsL gene which is specific to the Rev1 vaccine strain (21), was also checked by KMA. One more primer pair, which was found to be specific for B. melitensis in Kazakhstan (Forward_Bm and Reverse_Bm-r), was included as well (22). The in-silico polymerase chain reaction (PCR) was performed in Ugene (23) using Bruce-ladder multiplex PCR primers for determining the species (24). The product sizes of our five strains and 10 strains described by García-Yoldi et al (24) were imported to BioNumerics, and a dendrogram was plotted accordingly. For the Kmer-based identification of the bacterial strain, KmerFinder tools were used from the CGE website by submitting the draft assembly to the portal (https://cge.cbs.dtu.dk/services/KmerFinder/).
Table 1.
The Primers’ Sequences Used for In Silico PCR
|
|
Sequences
|
|
B. melitensis-specific primers
|
|
| IS711 - Forward |
5’- AAATCGCGTCCTTGCTGGTCTGA -3’ |
| IS711 - Reverse |
5’- TGCCGATCACTTAAGGGCCTTCAT -3’ |
| Vaccine strain Rev 1-specific primers |
|
| rpsL Rev1-Forward |
5’- CAGGCAAACCCTCAGAAGC -3’ |
| rpsL Rev1-Reverse |
5’- GATGTGGTAACGCACACCAA -3’ |
|
B. abortus and B. melitensis differentiating primers
|
|
| UF1 |
5’- GGCTATCGGCTGGGAAAGG -3’ |
| UR1 |
5’- CCTTCCGAAGAAAATACCCCT -3’ |
|
B. melitensis-specific primers for strains from Kazakhstan
|
|
| Forward_Bm |
5’- TCCAAACGCTTTCCCGGACGA -3’ |
| Reverse_Bm-r |
5’- GGCGAAACGGAAAAAGGTATCTCCAC -3’ |
Note. B. melitensis: Brucella melitensis; B. abortus: Brucella abortus; PCR: Polymerase chain reaction.
Phage Sequence and Antibiotic Resistance Genes
To check for the presence of the possible phage sequences, draft assemblies were submitted to PHASTER (http://phaster.ca/), and to predict the antibiotic resistome, Resistance Gene Identifier (RGI) (https://card.mcmaster.ca/analyze/rgi) was employed with the default parameters.
Virulence Factors
For the identification of virulence factors, all bacterial virulence factors were downloaded from the virulence factor database (25), and virulence proteins for B. melitensis bv.1 str. 16M (henceforth ‘16M strain’) were extracted from this database. The database contains 51 virulence factors, of which 31, 11, 6, and 2 cases belong to immune evasion, the secretion system, iron regulation, and iron uptake, respectively, and one case was for intracellular survival (Table 2). A blast database was created (using the makeblastdb command) from these proteins. The protein FASTA files of our strains were compared against this blast database by applying the blastp command with outfmt 6. The output format was filtered to keep the best score match for each protein in the database.
Table 2.
The Virulence Factors (n = 51) in Brucella melitensis bv 1 str. 16M
|
LPS (Immune Evasion)
|
CbetaG (Intracellular Survival)
|
| AcpXL |
BMEI1111;
BMEI1475
|
Cgs |
BMEI1837 |
| FabZ |
BMEI0832 |
|
|
| Gmd |
BMEI1413 |
Brucebactin (Iron uptake) |
| HtrB |
BMEI1115 |
DhbA |
BMEII0080 |
| KdsA |
BMEI0850 |
DhbB |
BMEII0079 |
| KdsB |
BMEI1904 |
DhbC |
BMEII0077 |
| LpsA |
BMEI1326 |
DhbE |
BMEII0078 |
| LpsB/LpcC |
BMEI0509 |
EntD |
BMEII0081 |
| LpxA |
BMEI0833 |
VibH/EntF |
BMEII0076 |
| LpxB |
BMEI0835 |
|
|
| LpxC |
BMEI0586 |
BvrRS (Regulation) |
| LpxD |
BMEI0831 |
BvrR |
BMEI2036 |
| LpxE |
BMEI1212 |
BvrS |
BMEI2035 |
| LpxK |
BMEII1028 |
|
|
| ManAoAg |
BMEI1394 |
Type IV secretion system |
| ManBcore |
BMEII0899 |
VirB1 |
BMEII0025 |
| ManCcore |
BMEII0900 |
VirB2 |
BMEII0026 |
| ManCoAg |
BMEI1395 |
VirB3 |
BMEII0027 |
| Per |
BMEI1414 |
VirB4 |
BMEII0028 |
| Pgm |
BMEI1886 |
VirB5 |
BMEII0029 |
| Pmm |
BMEI1396 |
VirB6 |
BMEII0030 |
| WaaA/kdtA |
BMEII1029 |
VirB7 |
BMEII0031 |
| WbdA |
BMEI0997 |
VirB8 |
BMEII0032 |
| WbkA |
BMEI1404 |
VirB9 |
BMEII0033 |
| WbkB |
BMEI1417 |
VirB10 |
BMEII0034 |
| WbkC |
BMEI1418 |
VirB11 |
BMEII0035 |
| WboA |
BMEI0998 |
|
|
| WbpL |
BMEI1426 |
|
|
| WbpZ |
BMEI1393 |
|
|
| Wzm |
BMEI1415 |
|
|
| Wzt |
BMEI1416 |
|
|
Note. Note. B. melitensis: Brucella melitensis.
Multilocus Sequence Typing
Two MLST schemes, 9 loci (gap, aroA, glk, dnaK, gyrB, trpE, cobQ, int_hyp, & omp25) (26) and 21 (gap, aroA, glk, dnaK, gyrB, trpE, cobQ, int_hyp, omp25, prpE, caiA, csdB, soxA, leuA, mviM, fumC, fbaA, ddlA, putA, mutL, & acnA) (26) loci were used for the typing of the strains. MLST genes were downloaded from PubMLST (26) and applied with the mlst program downloaded from GitHub (https://github.com/tseemann/mlst) to create databases and identify alleles and sequence types. Allelic combinations for which sequence type information was unavailable were then submitted to the PubMLST database for the subtype assignment with the ‘exact or nearest match option’. For MLST-based phylogenetic analysis, the nucleotide sequences of alleles were extracted by the KMA program for both 9 genes and 21 genes for MLST. These alleles were imported to BioNumerics, and alignment was executed using default multiple alignments. A UPGMA tree was drawn from this alignment using Kimura correction.
Single-Nucleotide Polymorphism-Based Phylogenetic Analysis
The nucleotide FASTA files of all strains from other countries were simulated to FASTQ file using Wgsim without introducing any mutation (with options -e 0.000 -r 0.0000 -R 0.00 -X 0.00). All these FASTQ reads, along with the FASTQ reads of the Kuwaiti strains, were imported to BioNumerics software and mapped against B. melitensis 16M using the default algorithm. The mapped reads were then imported to the wgSNP pipeline of the software to find genome-wide SNPs. Only high-quality SNPs with at least 12 base differences were extracted to draw the phylogenetic tree. From this SNP information, a neighbor-joining tree based on SNP values was drawn by rooting at B. abortus.
Results
Brucella melitensis-Specific Genetic Marker Identification in Kuwaiti Isolates
The IS711 marker was found in all five Kuwaiti isolates but of two different sizes (i.e., 732 and 733 bases), the details of which are depicted in Figure 1. All the Kuwaiti samples were negative for the rpsL gene and B. melitensis-specific primer product for Kazakhstan. A product of 84 bases, which is specific for B. melitensis, was obtained with all strains through primer pair UF1 and UR1 (Figure 2). For Bruce-ladder multiplex in-silico PCR, the band pattern in all five strains was the same with six bands of different sizes (152, 450, 587, 794, 1071, and 1862 bases, Figure 3), which is a characteristic band pattern for non-vaccine B. melitensis strains. A dendrogram with a comparison to other Brucella species is also shown in Figure 3.

Figure 1.
Multiple Sequence Alignment of IS711 Sequence of Brucella melitensis 16M With Kuwaiti Strains. The shaded area illustrates the similarity, while the white boxes represent nucleotide differences
.
Multiple Sequence Alignment of IS711 Sequence of Brucella melitensis 16M With Kuwaiti Strains. The shaded area illustrates the similarity, while the white boxes represent nucleotide differences

Figure 2.
Multiple Sequence Alignment of UF1 and UR1 Primer Product of Brucella melitensis 16M With Kuwaiti Strains
.
Multiple Sequence Alignment of UF1 and UR1 Primer Product of Brucella melitensis 16M With Kuwaiti Strains

Figure 3.
In Silico Multiplex Polymerase Chain Reaction. Note. PCR: Polymerase chain reaction. The length of the gene product was obtained using Ugene in silico PCR, and the dendrogram was constructed in BioNumerics using the known band pattern of Brucella species to compare the band pattern of Kuwaiti isolates
.
In Silico Multiplex Polymerase Chain Reaction. Note. PCR: Polymerase chain reaction. The length of the gene product was obtained using Ugene in silico PCR, and the dendrogram was constructed in BioNumerics using the known band pattern of Brucella species to compare the band pattern of Kuwaiti isolates
Phage Sequence and Resistance Genes
The sequence of an intact phage (Paraco vB PmaS IMEP1 of 13.6 kb length) was detected in all five genomes of Kuwaiti isolates with a score of 130. An additional phage Pseudo MD8 was also found in one of the genomes (i.e., ku_rcf_84). The output of RGI, when checked for the protein homolog model, suggested the presence of two antibiotic-resistance genes. The first one is B. suis mprF (AMR gene family: Defensin-resistant MprF), which belongs to the peptide antibiotic class and helps in antibiotic target alteration. The second one is adeF (AMR gene family: Resistance-nodulation-cell division antibiotic efflux pump), which belongs to the drug class tetracycline and fluoroquinolone, and the resistance mechanism is by the antibiotic efflux.
Virulence Factors
All 51 virulence factors present in the database were found in the samples with at least 99% similarity with the 16M strain. The maximum number of mutations were found in the cyclic beta 1-2 glucan synthetase gene (data are unavailable).
Multilocus Sequence Typing
The MLST results demonstrated that there was no MLST type or subtype which was prevalent in the Mediterranean region, while subtype 8 was the most common among all the strains (Figures 4a and 4b). However, the resolution of 21 genes MLST was better than 9 genes MLST (Figures 4a and 4b).

Figure 4.
Multiple Sequence Alignment-based Phylogenetic Tree: (a) MLST 9 Gene Scheme and (b) MLST 21 Gene Scheme. Note. The UPGMA tree was generated by aligning 9 MLST and 21 MLST genes in Bionumerics. The country of origin and sequence type for the corresponding sample is shown in front of the strain
.
Multiple Sequence Alignment-based Phylogenetic Tree: (a) MLST 9 Gene Scheme and (b) MLST 21 Gene Scheme. Note. The UPGMA tree was generated by aligning 9 MLST and 21 MLST genes in Bionumerics. The country of origin and sequence type for the corresponding sample is shown in front of the strain
Whole-Genome Single-Nucleotide Polymorphism-Based Phylogenetic Analysis
The wgSNP-based phylogenetic tree revealed that out of the five Kuwaiti strains, four strains have proximity with the strains from Iran, Iraq, Jordan, Afghanistan, and Turkmenistan, which are geographically in the north-east region of Kuwait, while one strain was found highly close to the strain from Somalia, which is located in the south of Kuwait (Figure 5).
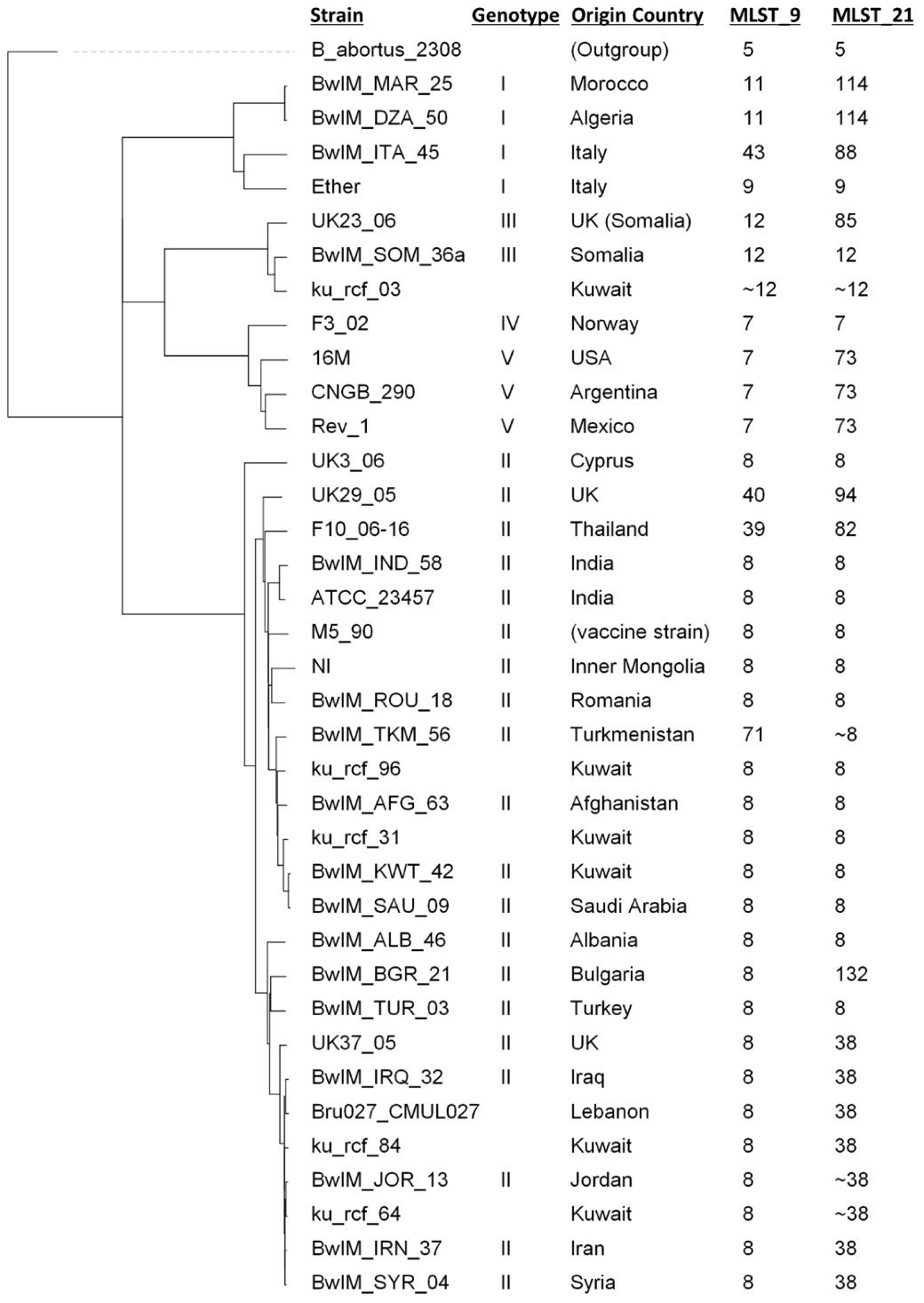
Figure 5.
wgSNP-based Phylogenetic Tree Comparing 32 Brucella Isolates From NCBI With 5 Kuwaiti Isolates. Note. wgSNP: Whole-genome single-nucleotide polymorphism; NCBI: National Center for Biotechnology Information. The tree was generated in BioNumerics using wgSNP data and rooted at B. abortus. The corresponding information such as genotype (as mentioned in previous studies [12,17,18]), country of origin, and sequence type through 9 and 21 gene schemes is presented in front of the strain name
.
wgSNP-based Phylogenetic Tree Comparing 32 Brucella Isolates From NCBI With 5 Kuwaiti Isolates. Note. wgSNP: Whole-genome single-nucleotide polymorphism; NCBI: National Center for Biotechnology Information. The tree was generated in BioNumerics using wgSNP data and rooted at B. abortus. The corresponding information such as genotype (as mentioned in previous studies [12,17,18]), country of origin, and sequence type through 9 and 21 gene schemes is presented in front of the strain name
Discussion
Brucellosis is considered endemic in most countries of the Middle East and poses a huge burden on humans and livestock (2,3). B. melitensis is the main cause of human brucellosis in the Middle East (4,27). There is a need for the investigation of B. melitensis genomes from the Middle East as the brucellosis cases in several non-endemic countries in the world are linked to the Middle East (17,28). This work is the continuation of our work on five draft genome sequences of B. melitensis (15). This study further investigated these genomes to identify several genomic markers related to species identification, pathogenesis, antibiotic resistance, and virulence.
Different markers can be used for the identification of Brucella species (9). In this study, we have taken insertion sequence IS711, a product of primer pair UF1-UR1, and a combination of eight primers of Bruce ladder multiplex PCR. All these markers were present in our isolates, which were identified as B. melitensis. Another gene rpsL that can differentiate between the vaccine and non-vaccine strains was absent from all the samples. All these primers were used for the in-silico PCR, where the template and primers were given in FASTA file format. This indicates that these techniques can be utilized in the routine identification of species. We also applied Kmerfinder, a web program, which can be used for the identification of species from FASTQ files, and confirmed that the species was B. melitensis.
Phage sequences, identified by PHASTER, were Paracoccus phage vB_PmaS-R3 in all the samples. This phage propagates through lytic mode and belongs to the viral family Siphoviridae (29). The genome of phage vB_PmaS-R3 has been divided into three regions, including genes for DNA metabolism, regulatory genes, and structure-forming genes (29). In addition, four gene transfer agent-like genes are present in the vB_PmaS-R3 phage, a putative L-alanyl-D-glutamate peptidase is predicted as the endolysin, and a mazG gene is found in the vB_PmaS-R3 phage indicating adaption to the nutrient-limited environment (29). The other phage was Pseudomonas phage MD8, which was found only in one of the samples. However, Paenibacillus phage Tripp was found in the 16M strain, which was considered a reference strain in this study. According to the previous study in Lebanon, the phages found in those samples were Brucella phage BiPBO1 and Lactococcus phage jj50 (18). However, none of these phages were identified in our samples. It will be interesting to know if these phages have any role in the pathogenic mechanisms of B. melitensis.
The lack of horizontal transfer in B. melitensis due to strict intracellular niche makes mutations, including point mutations and structural rearrangements, the exclusive heritable elements. These changes indicate the acquired adaptation upon introduction to the new geographical region (30). In the current study, a wgSNP-based approach using previously classified strains was considered to infer the phylogeny of the strains isolated from Kuwait. According to the phylogenetic tree, four out of five strains had a close relationship with the Eastern Mediterranean strains, but one of the strains was found close to a Somali strain which falls in the African clade. Additionally, wgSNP was compared against traditional MLST typing, and it was concluded that wgSNP has far better resolution in differentiating strains that were clubbed together through MLST.
It was also found that all the virulence factors present in the 16M strain were also present in Kuwaiti B. melitensis strains but with few mutations. The reason for these mutations could be environmental stress or the introduction of the Brucella vaccine (21). Some of these virulence factors were absent in studies performed with Brucella melitensis from other countries of the Middle East (18,21). This further suggests the importance of WGS in regular surveillance of infectious diseases.
In our study, we have affirmed the idea that wgSNPs can be exploited to identify subtypes or genotypes prevalent in specific geographical locations using the previously identified strains. This also provides information about the history and origin of the organism and its spread. B. melitensis has shown a 5-30% relapse rate in treated subjects (9), and due to high genomic similarity among Brucella species and strains, it is often extremely difficult to differentiate between relapse and reinfection (9). In this scenario, WGS can be highly efficient as other routinely used techniques cannot be differentiated at the genotypic/subtypic level. In addition, genotypic identification can also inform about the possible source in the case of biological warfare (31). In this study, we have described several tools which can be exploited to find the species from the draft genome. Some of these tools are specific for Brucella while others such as KmerFinder and in-silico PCR can be used generally to find species in any de novo assembly. Applying any of these techniques is far easier and more economical once obtaining the draft genome. Furthermore, it is important to have these genome sequences rather than storing DNA, which can be employed to identify other markers, if discovered in the future, which can be the markers of biotyping, vaccine strains, or antibiotic resistance genes. The low cost and high speed of WGS have practical implications in the routine diagnoses of microbial diseases (32), and it has shown the potential to replace the traditional gold standard typing methods such as PFGE, MLST, MLVA, 16S rRNA gene sequencing, and multiplex PCR, and the like (33).
Conclusion
In-silico comparative genome analysis is a fast and reliable tool to identify Brucella species and strains. Whole-genome analysis can differentiate between strains to an extent not possible by classical molecular methods. Furthermore, it can provide data about variance in genetic elements, the existence of drug-resistance genes, and SNP-based phylogeny. To the best of our knowledge, this is the first in-depth study of the isolates from Kuwait and their comparison with strains from some other Middle Eastern countries. Combined with the other studies of isolates from nearby regions, this can prove to be an effective tool in connecting the dots of missing information about B. melitensis infection prevalent in the Middle East. We have also demonstrated the use of several in-silico or web-based tools which are easy to implement and efficient in providing information about species and genotypes.
Authors’ Contribution
Conceptualization: Abu Salim Mustafa.
Data curation: Abu Salim Mustafa, Mohd Wasif Khan.
Formal analysis: Abu Salim Mustafa, Mohd Wasif Khan.
Funding acquisition: Abu Salim Mustafa.
Investigation: Abu Salim Mustafa, Nazima Habibi.
Methodology: Nazima Habibi, Mohd Wasif Khan.
Project administration: Abu Salim Mustafa.
Resources: Abu Salim Mustafa.
Supervision: Abu Salim Mustafa.
Validation: Abu Salim Mustafa, Mohd Wasif Khan.
Visualization: Abu Salim Mustafa, Nazima Habibi.
Writing–original draft: Abu Salim Mustafa, Mohd Wasif Khan.
Writing–review & editing: Abu Salim Mustafa, Mohd Wasif Khan, Nazima Habibi.
Competing Interests
None.
Ethical Approval
The Ethical Committee of Health Sciences, Kuwait University approved the investigation (the ethical approval No: RM01/13).
Funding
The study was supported by Kuwait University Research grants RM01/13 and SRUL02/13.
References
- Silva TM, Costa EA, Paixão TA, Tsolis RM, Santos RL. Laboratory animal models for brucellosis research. J Biomed Biotechnol 2011; 2011:518323. doi: 10.1155/2011/518323 [Crossref] [ Google Scholar]
- Hull NC, Schumaker BA. Comparisons of brucellosis between human and veterinary medicine. Infect Ecol Epidemiol 2018; 8(1):1500846. doi: 10.1080/20008686.2018.1500846 [Crossref] [ Google Scholar]
- Bagheri Nejad R, Krecek RC, Khalaf OH, Hailat N, Arenas-Gamboa AM. Brucellosis in the Middle East: current situation and a pathway forward. PLoS Negl Trop Dis 2020; 14(5):e0008071. doi: 10.1371/journal.pntd.0008071 [Crossref] [ Google Scholar]
- Al-Nakkas A, Mustafa AS, Wright SG. Large-scale evaluation of a single-tube nested PCR for the laboratory diagnosis of human brucellosis in Kuwait. J Med Microbiol 2005; 54(8):727-30. doi: 10.1099/jmm.0.45772-0 [Crossref] [ Google Scholar]
- Rossetti CA, Arenas-Gamboa AM, Maurizio E. Caprine brucellosis: a historically neglected disease with significant impact on public health. PLoS Negl Trop Dis 2017; 11(8):e0005692. doi: 10.1371/journal.pntd.0005692 [Crossref] [ Google Scholar]
- Wang H, Xu WM, Zhu KJ, Zhu SJ, Zhang HF, Wang J. Molecular investigation of infection sources and transmission chains of brucellosis in Zhejiang, China. Emerg Microbes Infect 2020; 9(1):889-99. doi: 10.1080/22221751.2020.1754137 [Crossref] [ Google Scholar]
- Pelerito A, Nunes A, Núncio MS, Gomes JP. Genome-scale approach to study the genetic relatedness among Brucella melitensis strains. PLoS One 2020; 15(3):e0229863. doi: 10.1371/journal.pone.0229863 [Crossref] [ Google Scholar]
- Zheludkov MM, Tsirelson LE. Reservoirs of Brucella infection in nature. Biology Bulletin 2010; 37(7):709-15. doi: 10.1134/s106235901007006x [Crossref] [ Google Scholar]
- Mustafa AS, Habibi N, Osman A, Shaheed F, Khan MW. Species identification and molecular typing of human Brucella isolates from Kuwait. PLoS One 2017; 12(8):e0182111. doi: 10.1371/journal.pone.0182111 [Crossref] [ Google Scholar]
- Liu ZG, Wang M, Zhao HY, Piao DR, Jiang H, Li ZJ. Investigation of the molecular characteristics of Brucella isolates from Guangxi Province, China. BMC Microbiol 2019; 19(1):292. doi: 10.1186/s12866-019-1665-6 [Crossref] [ Google Scholar]
- Oniciuc EA, Likotrafiti E, Alvarez-Molina A, Prieto M, Santos JA, Alvarez-Ordóñez A. The present and future of whole genome sequencing (WGS) and whole metagenome sequencing (WMS) for surveillance of antimicrobial resistant microorganisms and antimicrobial resistance genes across the food chain. Genes (Basel) 2018; 9(5):268. doi: 10.3390/genes9050268 [Crossref] [ Google Scholar]
- Tan KK, Tan YC, Chang LY, Lee KW, Nore SS, Yee WY. Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genomics 2015; 16(1):93. doi: 10.1186/s12864-015-1294-x [Crossref] [ Google Scholar]
- Sankarasubramanian J, Vishnu US, Gunasekaran P, Rajendhran J. A genome-wide SNP-based phylogenetic analysis distinguishes different biovars of Brucella suis. Infect Genet Evol 2016; 41:213-7. doi: 10.1016/j.meegid.2016.04.012 [Crossref] [ Google Scholar]
- Dimitrov T, Panigrahi D, Emara M, Awni F, Passadilla R. Seroepidemiological and microbiological study of brucellosis in Kuwait. Med Princ Pract 2004; 13(4):215-9. doi: 10.1159/000078319 [Crossref] [ Google Scholar]
- Khan MW, Habibi N, Shaheed F, Mustafa AS. Draft genome sequences of five clinical strains of Brucella melitensis isolated from patients residing in Kuwait. Genome Announc 2016; 4(6):e01144-16. doi: 10.1128/genomeA.01144-16 [Crossref] [ Google Scholar]
- Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2016; 44(D1):D67-72. doi: 10.1093/nar/gkv1276 [Crossref] [ Google Scholar]
- Georgi E, Walter MC, Pfalzgraf MT, Northoff BH, Holdt LM, Scholz HC. Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS One 2017; 12(4):e0175425. doi: 10.1371/journal.pone.0175425 [Crossref] [ Google Scholar]
- Abou Zaki N, Salloum T, Osman M, Rafei R, Hamze M, Tokajian S. Typing and comparative genome analysis of Brucella melitensis isolated from Lebanon. FEMS Microbiol Lett 2017; 364(19):fnx199. doi: 10.1093/femsle/fnx199 [Crossref] [ Google Scholar]
- Nayeri Fasaei B, Etemadi A, Esmaelizad M, Zahraei Salehi T, Aghaiipoor K, Alamian S. Detection of Brucella melitensis and Brucella abortus strains using a single-stage PCR method. Arch Razi Inst 2015; 70(1):51-5. doi: 10.7508/ari.2015.01.008 [Crossref] [ Google Scholar]
- Clausen P, Aarestrup FM, Lund O. Rapid and precise alignment of raw reads against redundant databases with KMA. BMC Bioinformatics 2018; 19(1):307. doi: 10.1186/s12859-018-2336-6 [Crossref] [ Google Scholar]
- Awwad E, Adwan K, Farraj M, Essawi T, Rumi I, Manasra A. Cell envelope virulence genes among field strains of Brucella melitensis Isolated in West Bank part of Palestine. Agric Agric Sci Procedia 2015; 6:281-6. doi: 10.1016/j.aaspro.2015.08.073 [Crossref] [ Google Scholar]
- Daugaliyeva A, Peletto S, Sultanov A, Baramova S, Acutis PL, Adambaeva A. Development of a differential PCR assay for detection of Brucella abortus and Brucella melitensis: an analytical approach for monitoring of Brucella spp in foods of animal origin. J Food Qual Hazards Control 2016; 3(2):53-9. [ Google Scholar]
- Okonechnikov K, Golosova O, Fursov M. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 2012; 28(8):1166-7. doi: 10.1093/bioinformatics/bts091 [Crossref] [ Google Scholar]
- García-Yoldi D, Marín CM, de Miguel MJ, Muñoz PM, Vizmanos JL, López-Goñi I. Multiplex PCR assay for the identification and differentiation of all Brucella species and the vaccine strains Brucella abortus S19 and RB51 and Brucella melitensis Rev1. Clin Chem 2006; 52(4):779-81. doi: 10.1373/clinchem.2005.062596 [Crossref] [ Google Scholar]
- Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res 2005; 33(Database issue):D325-8. doi: 10.1093/nar/gki008 [Crossref] [ Google Scholar]
- Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLSTorg website and their applications. Wellcome Open Res 2018; 3:124. doi: 10.12688/wellcomeopenres.14826.1 [Crossref] [ Google Scholar]
- Al Nakkas AF, Wright SG, Mustafa AS, Wilson S. Single-tube, nested PCR for the diagnosis of human brucellosis in Kuwait. Ann Trop Med Parasitol 2002; 96(4):397-403. doi: 10.1179/000349802125001203 [Crossref] [ Google Scholar]
- Garofolo G, Fasanella A, Di Giannatale E, Platone I, Sacchini L, Persiani T. Cases of human brucellosis in Sweden linked to Middle East and Africa. BMC Res Notes 2016; 9:277. doi: 10.1186/s13104-016-2074-7 [Crossref] [ Google Scholar]
- Xu Y, Zhang R, Jiao N. Complete genome sequence of Paracoccusmarcusii phage vB_PmaS-R3 isolated from the South China Sea. Stand Genomic Sci 2015; 10:94. doi: 10.1186/s40793-015-0089-7 [Crossref] [ Google Scholar]
- Moreno E. Genome evolution within the alpha Proteobacteria: why do some bacteria not possess plasmids and others exhibit more than one different chromosome?. FEMS Microbiol Rev 1998; 22(4):255-75. doi: 10.1111/j.1574-6976.1998.tb00370.x [Crossref] [ Google Scholar]
- Doganay GD, Doganay M. Brucella as a potential agent of bioterrorism. Recent Pat Antiinfect Drug Discov 2013; 8(1):27-33. doi: 10.2174/1574891x11308010006 [Crossref] [ Google Scholar]
- Rossen JWA, Friedrich AW, Moran-Gilad J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect 2018; 24(4):355-60. doi: 10.1016/j.cmi.2017.11.001 [Crossref] [ Google Scholar]
- Tang S, Orsi RH, Luo H, Ge C, Zhang G, Baker RC. Assessment and comparison of molecular subtyping and characterization methods for Salmonella. Front Microbiol 2019; 10:1591. doi: 10.3389/fmicb.2019.01591 [Crossref] [ Google Scholar]