Avicenna Journal of Clinical Microbiology and Infection. 8(2):66-73.
doi: 10.34172/ajcmi.2021.12
Original Article
Reverse Vaccinology Approach for a Potential Rhinovirus Vaccine
Shuaibu Abdullahi Hudu 1, *  , Saadatu Haruna Shinkafi 2, Shuaibu Umar 2, Babazhitsu Makun 1
, Saadatu Haruna Shinkafi 2, Shuaibu Umar 2, Babazhitsu Makun 1
Author information:
1Department of Medical Microbiology and Parasitology, Faculty of Basic Clinical Sciences, College of Health Sciences, Usmanu Danfodiyo University, Sokoto, 840232 Sokoto State, Nigeria
2Department of Microbiology and Parasitology, Usmanu Danfodiyo University Teaching Hospital Sokoto, 80002 Sokoto State, Nigeria
*
Corresponding author: Shuaibu Abdullahi Hudu Department of Medical Microbiology and Parasitology, Faculty of Basic Clinical Sciences, College of Health Sciences, Usmanu Danfodiyo University, Sokoto, 840232 Sokoto State, Nigeria. Email:
hudu.shuaibu@udusok.edu.ng
Abstract
Background: Rhinoviruses (RVs) represent the most important aetiological agents of the common cold and are responsible for about two-thirds of acute exacerbations of chronic bronchitis, asthma, and chronic obstructive pulmonary disease (COPD) in both children and adults. This study aimed to design a pan-serotypic vaccine capable of inducing cross-reactive antibodies against most of the RV by using a reverse Vaccinology approach.
Methods: Bioinformatics analysis was carried out to characterise the capsid proteins (VP1, VP2, VP3, and VP4) of all known RV serotypes and to predict potential immune motifs. Conserved motifs consisting at least nine-mers common across all RV-A or B serotypes were selected and synthesized chemically. Four tagged full-length genes coding the capsid proteins of an ideal strain (HRV-74), VP1, VP2, VP3, and VP4 were constructed and cloned in vitro. Upon expression, the purified recombinant proteins were also administered subcutaneously to other groups of rabbits. The responses and cross-reactivity of the specific immunoglobulin M (IgM) and G (IgG) to the peptides, proteins, and whole viruses were measured.
Results: The obtained anti-peptide antibodies exhibited a cross-neutralizing activity for different RV strains in vitro. Antibodies raised to the synthetic peptides exhibited cross-reactivity against the corresponding recombinant proteins and antigenically distinct RV strains coated on plates via ELISA assay.
Conclusions: The study findings indicated that the peptides corresponding to the conserved region of the RV capsid proteins were potent immunogenic; moreover, the findings showed that their combination was crucial for extending the cross-protection against variant RVs.
Keywords: Antibodies, Cross-protection, Reverse vaccinology, Rhinovirus, Vaccine
Copyright and License Information
© 2021 The Author(s); Published by Hamadan University of Medical Sciences.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
http://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium provided the original work is properly cited.
Background
Rhinovirus (RV) was first isolated by Pelon et al and Price in 1956 (1). It is considered as the most important cause of common cold and is well recognised as causative agents for the self-limiting disease of the respiratory tract. The use of advanced diagnostic methods in a wide variety of studies has demonstrated that RVs are also associated with the severity of respiratory symptoms. According to several reports, RVs are responsible for about 75% of acute asthma exacerbations and chronic obstructive pulmonary disease (COPD) in both children and adults (2-5) pneumonia (6), acute bronchiolitis in young children (7), croup (8) and otitis media (9). The number of cases is growing globally in each consecutive year, which increases the burden of providing care worldwide (10). The RVs genomic RNA is surrounded by a viral capsid which is a structural protein made up of four proteins designated as VP1, VP2, VP3 and VP4; out of these four proteins, VP1, VP2 and VP3 are larger proteins that are exposed to the capsid surface, thereby accounting for the viral antigenic diversity. VP4, on the other hand, is a small protein that lies between the caps and viral genome as such, and less antigenic. It is almost impossible to avoid RV infection since millions of these viral particles are transmitted via exhalation during conversation, sneezing, coughing, and direct contact; this virus can also remain infectious for a relatively long time (11).
To date, however, no effective antiviral therapy has been approved for either the prevention or the treatment of RVs infections. This might be due to the large number of RVs stereotypes with hypervariable sequence, the lack of an animal model, and the rapid emergence of new strains, all hindering the progression of vaccine development. Considering the high RV burden of infection, which is poorly responsive to the current therapies, an alternative approach is urgently needed. Therefore, this study aimed at developing a pan-serotype vaccine capable of inducing cross-reactive antibodies by using reverse vaccinology approach.
Methods
Conserved Region Sequence Identification
Protein sequences of all known distinct RV prototypes were downloaded from the National Centre for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov) in September 2019. However, HRV-87 was excluded from the analysis since it had been reclassified as a human enterovirus (12,13). VP1 data under GenBank accession numbers AY355180 to AY355281 described by Ledford et al (14) were considered to confirm the accuracy of the analysis. The obtained RV sequences were grouped based on their original classification as RV A (n=63), RV B (n=25), and RV C (n=12). After grouping, the sequence editing was carried out using Biological Sequence Alignment Editor, v 7.2.0 (BioEdit software) in order to study each capsid protein (VP1, VP2, VP3 and VP4) independently (15). Sequence alignments were performed for all available complete capsid proteins from both HRV-A and B by using MUSCLE multiple sequence alignment algorithm (16). Conserved regions that were common across all RV-A and B serotypes and exhibited at least 80% representation were selected. Similarly, selected sequences were further analysed using EMBL-EBI ClustalW server (The European Bioinformatics Institute; http://www.ebi.ac.uk) for calculation of the identities, similarities, and differences for the selected sequences which finally generated consensus sequences and distance matrix. A Phylogenetic tree of amino acid sequences for all capsid proteins was constructed using the neighbour-joining (NJ) method – a feature of the software MEGA 5 (17). Reliability of phylogenetic groupings was evaluated to determine the strength of the nodes by using the bootstrap test with 100 replications.
Cloning and Expression of VP1-4 Recombinant Proteins
The full lengths of RV recombinant capsid proteins (VP1, VP2, VP3, and VP4) that were capable of inducing an immune response was constructed. The codon usage of HRV-74 capsid full-length genes, VP1-4, was analysed with Bio Basic Inc.’s optimization software based on the E. coli codon preference, and the sequence was submitted to GenBank with accession number KP063328- KP063331. These full-length and codon-optimized VP1-4 sequences were then chemically synthesised by Bio Basic (Ontario, Canada), with addition six histidine residues (His-tag) at their N-termini so as to allow detection and purification of protein and restriction enzyme sites of NdeI and XhoI for sub-cloning purposes immediately upstream of the His-Tag sequence and downstream of the inserted stop codon, respectively. All the synthesized codon-optimized genes coding for VP1-4 were separately cloned into pET-21a (+) expression vector (Novagen, USA) and transformed into competent E. coli BL21 (DE3) cells (Novagen, USA) according to the manufacturer’s recommended protocol. After overnight growth in the presence of ampicillin, grown individual colonies were screened by PCR using published primers (18).
The PCR products were analysed by 1% agarose gel electrophoresis stained with GelRed (Biotium Inc., Hayward, CA, USA) and visualized by using the Gel Doc XR imager (Bio-Rad). Positive plasmids were purified via a PureLink HiPure Plasmid DNA Purification Kit (Invitrogen) according to manufacturer’s recommendations, and sequenced by First BASE Laboratories (Selangor, Malaysia).
Extraction and Purification of Recombinant Proteins
The cultures were harvested after centrifugation for 10 minutes at 5000 g,and the bacteria were suspended in 4 mL of B-PER lyses reagent (Thermo Scientific, Massachusetts, USA) until they were homogenised. The homogenised suspension was incubated for 15 minutes at room temperature (RT) and centrifuged at 15000 g for 5 minutes and then the supernatant and precipitate fractions were separated. Inclusion bodies were solubilised with inclusion body solubilisation reagent (Pierce, Rockford, USA), and the proteins were refolded according to the manufacturer’s protocol (19). Both soluble and insoluble fractions were stored in small aliquots at -80°C for further analysis.
The His-tagged recombinant protein (VP1, 2, 3, or 4) was purified using HisPur Cobalt Purification Kit according to manufacturer’s guideline. Briefly, each given column, pre-filled with 3 mL resin bed, was designated to one specific fusion protein plus negative control to prevent cross contamination of the samples. Mixture of the extracted proteins and Equilibration/Wash Buffer (v/v) was prepared and added to the cobalt resin in the Spin Column. Bound protein was eluted using one resin-bed volume of Elution Buffer (50mM sodium phosphate, 300mM sodium chloride, 150mM imidazole, at pH 7.4). The fractions were then stored at -80°C in small aliquots for further analysis. The prepared proteins were dialysed against buffers with decreasing urea concentration. The proteins were concentrated and dialysed as described previously by Fling and Gregerson (20) and Gagnon et al (21).
Protein Assays
The quantity, purity, and specificity of all purified recombinant proteins were checked using NanoDrop, SDS-PAGE, and Coomassie staining or western blot prior to the large scale production. Protein concentrations were determined using Pierce Coomassie Plus Bradford Protein Assay (Thermo Scientific, Rockford, USA) and following manufacturer’s recommendations. Briefly, 10 µL of each purified recombinant protein or standard was loaded into individual microplate wells. Then, 250-300 µL of the Coomassie plus Reagent was added to each well and mixed gently for 30 seconds. The plates were then incubated for 10 min at RT and read at an absorbance of 595 nm with a plate reader. A standard curve was constructed using the known concentrations of BSA. The generated curve was used to determine the concentration of each recombinant protein. Purified fractions were analysed by using SDS-PAGE (Precise gels; Thermo Scientific) to assess apparent protein purity according to the manufacturer’s instructions. To visualize the protein bands, gels were stained with SimplyBlue SafeStain from Invitrogen following the specified protocol. First, the gel was rinsed 3 times for 5 min each with about 100 µL of water. Then the gel was placed and shaken in about 20 mL of Simply-Blue SafeStain for 1 hour. Next, the gel was distained with about 100 µL of water for 1-2 hours and shaken at the same time. After confirming the band by SDS-PAGE gel, Western blot was carried out to confirm the presence of recombinant proteins using a Fast Western Blot kit with SuperSignal West Pico Substrate according to the manufacturer’s instructions (Pierce, USA).
Peptides Synthesis
The conserved motifs consisting at least nine-mers common across all RV-A were selected and synthesized chemically by Mimotopes (Clayton, Victoria, Australia). The purity of the peptides was >95%, as confirmed by high-performance liquid chromatography (HPLC). Once received, the lyophilised peptides were kept in -20ºC for further assays. Before using, however, each group of peptides corresponding to specific protein was diluted and mixed at the final required concentration. The purified recombinant proteins were also administered subcutaneously to other groups of Wistar rats. The responses and cross-reactivity of the specific immunoglobulin M (IgM) and G (IgG) to the peptides, proteins, and all viruses were measured.
Virus Propagation
As for immunoreactivity studies, the RV strains were propagated in H1-HeLa cells (ATCC, CRL-1958) two times before titrating and extracting the viral RNA as instructed in the ATCC product information sheet and passage. After reaching 80% confluence, the medium was removed and the cells were rinsed briefly with 5 mL PBS at RT. The monolayer was then infected with corresponding strain at a multiplicity of infection (MOI) of 0.1, as recommended by ATCC. Virus adsorption was carried out for approximately 1 hour at 37ºC in a humidified atmosphere in the presence of CO2 (5%) with a gentle manual rocking every 15 minutes. The virus-containing supernatant was collected and filtered through a 0.2 μm syringe filter and stored at -80ºC so that it would be used as a new stock of virus.
Rhinovirus Neutralization Assay In vitro
This test was performed in 96-well tissue culture flat-bottom plates as described previously (19). In sum, H1-HeLa cells (CRL-1958, ATCC) were seeded into wells and grown to 80-90% confluence. HRV-74 guinea pig antisera were purchased from ATCC (VR-1184 AS/GP), diluted as instructed in the ATCC product information sheet, and used as the positive control. For each experiment, cells with fresh medium were only used as negative control, while infected cells without antiserum were used as positive control. Minimum essential medium (MEM) Eagle medium (Biowest, Nuaillé, France) containing 2% FBS was used as a diluent in the experiments.
Cross-neutralization assays were also performed in 96-well plates, as described previously (22). The H1-HeLa cells were seeded in MEM containing 2% FBS and 1 mM glutamine (infection medium) and grown overnight at 37ºC to 80% confluency. RVs (HRV-A15, 16, 44, 51, 54, 55, 76, 89, and B72) (100 TCID50 in 100 µL infection medium) were mixed with 50 µL of the respective undiluted antiserum and serial two-fold dilutions thereof in the same medium. After incubating for 3 hours at 37ºC, the cells were overlaid with these solutions and the incubation was continued at 33ºC in a humidified 5% CO2 atmosphere for three days. The medium was removed and the cells were stained with 10% MEM of alamarBlue (Invitrogen Corp., CA, USA) and the plate were incubated for 2-4 hours, following the manufacturer’s protocol. On a plate reader (Sunrise Basic, Tecan, Austria), the plate was read at an absorbance of 570 nm with 600 nm as a reference wavelength. The cell viability was expressed as the cell survival ratio relative to the control (23). All the controls described above were applied for each cross-neutralizing test.
Results
Conserved RV Protein Region
Multiple alignments of VP1, VP2, VP3, and VP4 amino acid sequences showed several conserved regions across each selected group with a minimum length of 9 residues; and maximum entropy 0.2 was found. Three highly conserved region were identified for the VP1 upon multiple sequence alignment of all RV-A. On the other hand, RV-B showed no conservative motifs using the applied criteria. However, several conserved motifs within the group were observed once the maximum entropy changed to 0.5. A consensus sequence (305 aa) that represented the 75 aligned VP1 aa sequences was obtained. Further analysis showed that HRV-74 had the highest homology to consensus sequence (86%). This homology increased to 88.2% once we deleted the extra residues in consensus sequence (16 residues at five positions). The consensus identity within RV-A group ranged from a low level of 64.8% (HRV-8) to a high level of 86% (HRV-74) with median identity of 77.27%. It was also found that 101 residues (33.1% of the VP1 full length) at different positions were 100% conserved across all RV-A. As for VP2, three highly conserved region were also identified.
Unlike VP1, VP2 of RV-B exhibited six conserved motifs with several residues within the detected regions and appeared to be conserved across RV-A and B prototype strains. The consensus identity within RV-A group ranged from a low level of 74.7% (HRV-8) to a high level of 91.92% (HRV-21) with median identity of 84.7%. Moreover, the analysis indicated that 130 residues (48.14% of the full length) at different positions were 100% conserved across all RV-A. Within RV-B, consensus sequence (266 aa) of the 25 aligned VP2 aa sequences was generated.
The full-length of RV-A VP3 aa sequences was found to be variable from one strain to another and to vary in length from 236 aa residues of HRV-A30 and -A23 to 241 aa residues of HRV-A21. On the other hand, VP3 aa sequences of all RV-B serotypes showed no difference in sequences length which was 236 aa. Within RV-A, the level of VP3 amino acids identity ranged from a low level of 63.14% (HRV-30 and 8) to a high level of 100% (HRV-8 and 95), while RV-B amino acid sequence identity of the respective protein ranged from a low level of 71.61% (HRV-3 and 27) to a high level of 97.88% (HRV-70 and HRV-17). As for serotype pairs, 61 and 27 pairs had amino acid sequence identity of ≥ 90% for RV-A and -B, respectively. VP4 had the shortest sequence among other viral capsid proteins with only 69 aa residues in length. After multiple alignments, VP4 was found to be the most conserved protein among other proteins. Within RV-A, the level of VP4 aa identity ranged from a low level of 88.41% (several serotypes) to a high level of 100% (332 serotypes pairs), whereas RV-B amino acid sequence identity of the respective protein ranged from a low level of 88.41% (HRV-26 and 48) to a high level of 100% (5 serotype pairs). Compared to other capsid proteins, only 76 (2.7%) and 7 (2.3%) serotype pairs had amino acid sequence identity of less than 90% for RV-A and B, respectively.
Expressed Cloned VP 1-4 Recombinant Genes
All codon-optimized genes including VP1-4 that were separately cloned into pET-21a (+) expression vector were transformed successfully into competent E. coli BL21 (DE3) cells. After overnight incubation, the growth of single colonies was observed in the presence of ampicillin in culture medium, while no growth was detected in negative control plates. Furthermore, cloning and transformations were confirmed by colony PCR which demonstrated clear bands at the expected size for each protein gene (Figure 1). The recombinant proteins VP1, VP2, VP3, and VP4 were successfully expressed in E. coli and produced in large-scale. Molecular weight values of the recombinant proteins revealed in SDS-PAGE were 33.7 kDa, 30.1 kDa, 27.4 kDa, and 8.3 kDa for VP1, VP2, VP3, and VP4, respectively (Figure 2).
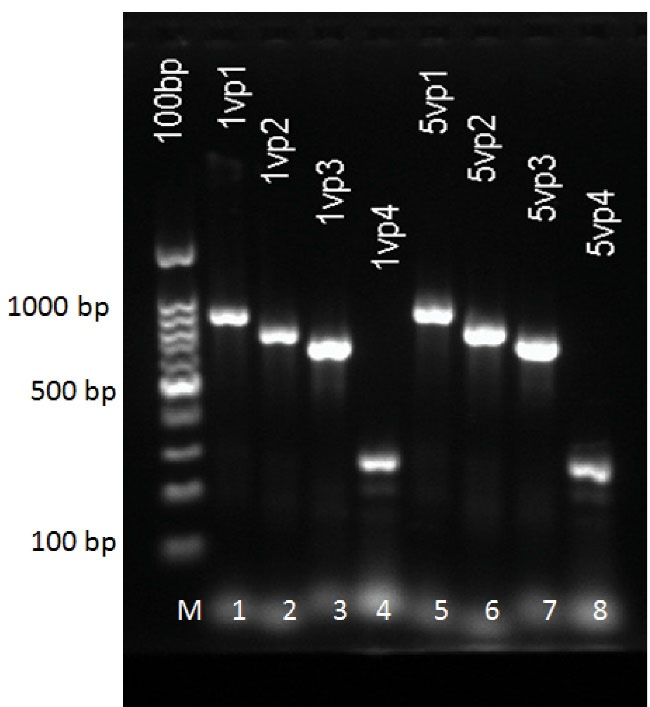
Figure 1.
Analysis of Two Single Colonies of Each Plate (4 plates) by PCR. Bands confirming the presence of the inserted fragments, VP1, VP2, VP3 and VP4. Lane 1 and 5: VP1; Lane 2 and 6: VP2; Lane 3 and 7: VP3; Lane 4 and 8: VP4; M: 100 bp DNA ladder.
.
Analysis of Two Single Colonies of Each Plate (4 plates) by PCR. Bands confirming the presence of the inserted fragments, VP1, VP2, VP3 and VP4. Lane 1 and 5: VP1; Lane 2 and 6: VP2; Lane 3 and 7: VP3; Lane 4 and 8: VP4; M: 100 bp DNA ladder.
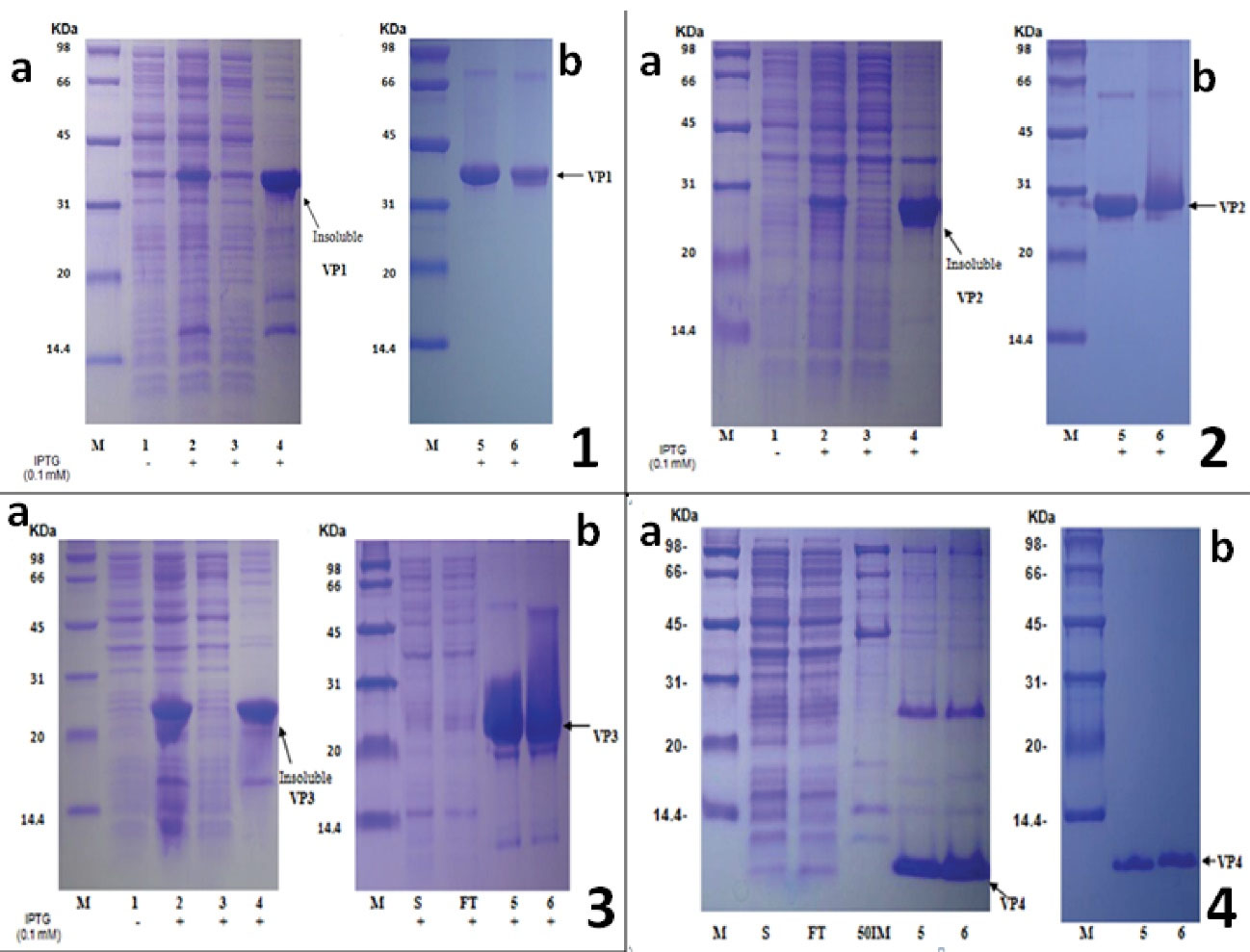
Figure 2.
SDS-PAGE Profile of E. coli BL21 Proteins Transformed With pET21a-VP1, VP2, VP3, VP4 and Their Corresponding Purified Recombinant Protein. Plate 1(a): protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP1 recombinant protein revealed by SDS-PAGE and ExPASy was 33.7 ± 3.3 kDa. (b): Purified recombinant VP1 protein by HisPur cobalt affinity chromatography and separated on 12% SDS-PAGE Gel. Plate 2 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP2 recombinant protein revealed by SDS-PAGE and ExPASy was 30.1±3.0 kDa. (b): Purified recombinant VP2 protein by HisPur cobalt affinity chromatography and separated on 12% SDS-PAGE gel. Plate 3 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP3 recombinant protein revealed by SDS-PAGE and ExPASy was 27.4±2.7 kDa. (b): Purification process of recombinant VP3 protein after dissolving the inclusion bodies with 20mM Tris-HCl, 300mM NaCl, 20mM imidazole, 20mM beta-ME, 7M GuaHCl, pH 8.0. Plate 4 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. No apparent target protein expression after IPTG induction. (b): Purification process of recombinant VP4 protein after sonication in 8M urea. The molecular mass of the recombinant VP4 protein revealed by SDS-PAGE and ExPASy was 8394±800 Da. M: Markers; Line 1: Control (No IPTG); Line 2 and WB: whole bacterial proteins; Line 3: supernatant after sonication; Line 4: precipitant after sonication; Line 5: reducing conditions; Line 6: non-reducing conditions; S: supernatant after dissolving by 7M GuaHCl; FT: Flow through. 50IM: Elution by 20mM Tris-HCl, 300mM NaCl, 50mM imidazole, 8M urea, pH8.0
.
SDS-PAGE Profile of E. coli BL21 Proteins Transformed With pET21a-VP1, VP2, VP3, VP4 and Their Corresponding Purified Recombinant Protein. Plate 1(a): protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP1 recombinant protein revealed by SDS-PAGE and ExPASy was 33.7 ± 3.3 kDa. (b): Purified recombinant VP1 protein by HisPur cobalt affinity chromatography and separated on 12% SDS-PAGE Gel. Plate 2 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP2 recombinant protein revealed by SDS-PAGE and ExPASy was 30.1±3.0 kDa. (b): Purified recombinant VP2 protein by HisPur cobalt affinity chromatography and separated on 12% SDS-PAGE gel. Plate 3 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. The molecular mass of the VP3 recombinant protein revealed by SDS-PAGE and ExPASy was 27.4±2.7 kDa. (b): Purification process of recombinant VP3 protein after dissolving the inclusion bodies with 20mM Tris-HCl, 300mM NaCl, 20mM imidazole, 20mM beta-ME, 7M GuaHCl, pH 8.0. Plate 4 (a): Protein expression was induced with 0.1mM IPTG at the cell density (OD600) of 0.5 to 0.6 and continued shaking for 3 h at 37°C. No apparent target protein expression after IPTG induction. (b): Purification process of recombinant VP4 protein after sonication in 8M urea. The molecular mass of the recombinant VP4 protein revealed by SDS-PAGE and ExPASy was 8394±800 Da. M: Markers; Line 1: Control (No IPTG); Line 2 and WB: whole bacterial proteins; Line 3: supernatant after sonication; Line 4: precipitant after sonication; Line 5: reducing conditions; Line 6: non-reducing conditions; S: supernatant after dissolving by 7M GuaHCl; FT: Flow through. 50IM: Elution by 20mM Tris-HCl, 300mM NaCl, 50mM imidazole, 8M urea, pH8.0
Purity of Extracted Proteins
All the expressed recombinant proteins were found as insoluble proteins produced in the inclusion bodies. These findings prompted us to solubilise the inclusion bodies by 20mM Tris-HCl, 300mM NaCl, 20mM Imidazole, 20mM beta-ME, 7M GuaHCl, and pH 8.0 in order to extract the desired proteins followed by purification step using HisPur Cobalt affinity chromatography. Western blot analyses indicated that the recombinant proteins VP1-4 reacted specifically with the rabbit anti-His6-tag antibody due to their N-terminal X6-histidine tag. After His-tag affinity purification, proteins concentrations and purity were determined. The expressed proteins were purified to single bands, as were confirmed by SDS-PAGE; the purity was between 85-95%.
Synthetic Peptides
Following bioinformatics analysis, three highly conserved regions were identified within each of the VP1 (ASAYYMFYDGY), VP2 (PYVNAVPMDSM), and VP4 (GSSLNYFNINYFKDAAS) protein sequences, while VP3 did not contain any well motif. These computationally identified regions among the RV-A capsid proteins were synthesized chemically by Mimotopes (Clayton, Victoria, Australia). The purity of the peptides was >95% as confirmed by HPLC. Peptides vary in length from 9 residues to 17 residues. The theoretical molecular weights of these peptides also varied from 913.94 to 1911.07.
In Vitro RV Neuralization
Nine variant RVs plus the ideal strain were successfully propagated in H1-HeLa cells (ATCC, CRL-1958). After the harvest, the viral identities were confirmed by RT-PCR; the PCR products that were purified, sequenced, and analysed showed 100% homologies to their corresponding serotypes. Neutralization assay was performed for the selected nine distinct serotypes. The results suggested that anti-VP1/VP4-proteins at different concentration had the highest neutralizing ability to all different serotypes at 100 TCID50 (Figure 3). In addition, all recombinant proteins demonstrated a good reactivity against almost all stereotypes, including HRV-72 from group B.
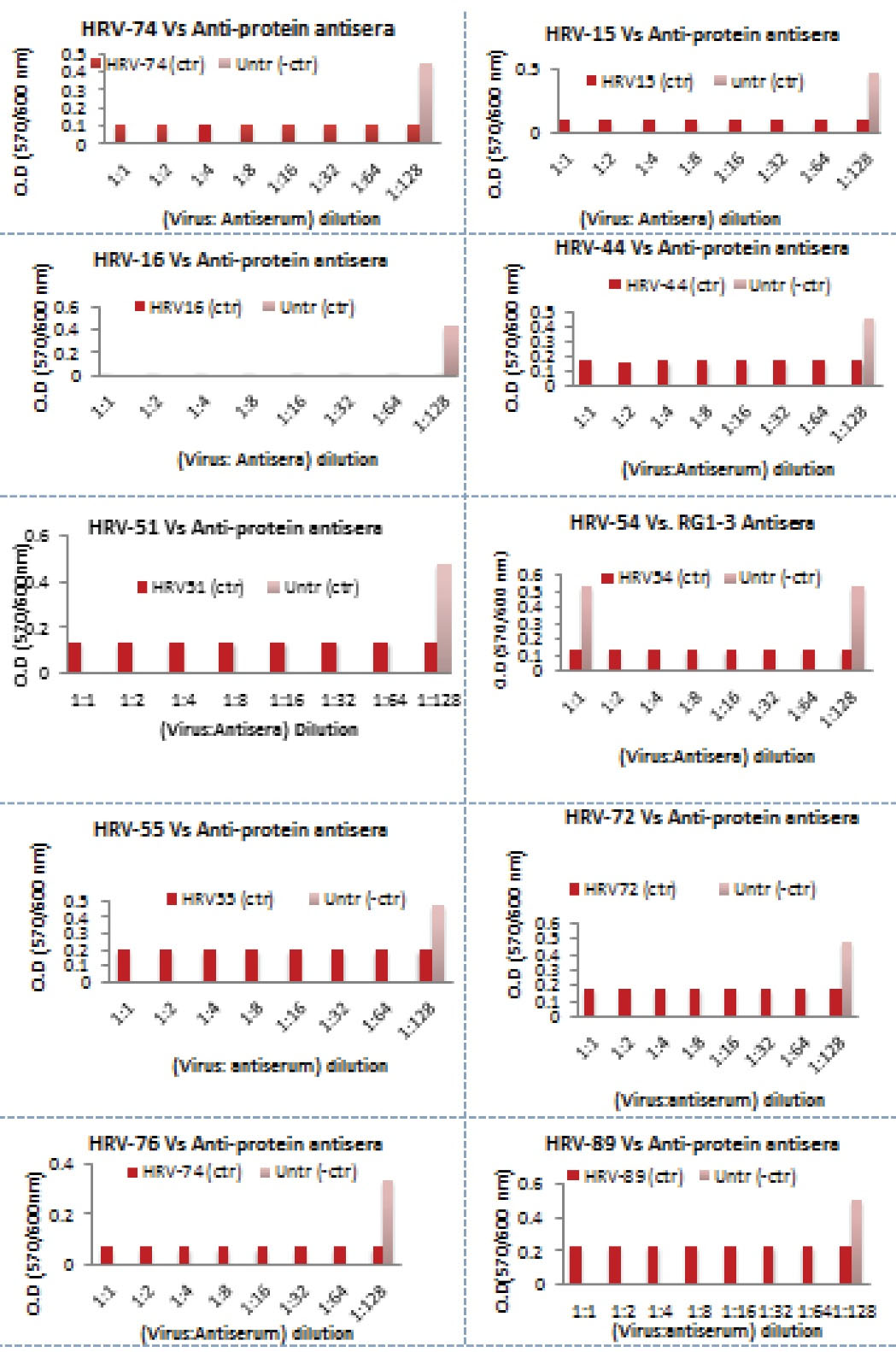
Figure 3.
Inhibition of HRV-74, 15, 16, 44, 51, 54, 55, 72, 76 and 89 Viral Infectivity (at 100 TCID50) by Elicited Specific Antibodies Raised Against Recombinant Capsid Proteins and Synthetic Peptides Corresponding to the Conserved Region of the RVs Capsid Proteins. Data are shown as mean ± SE.
.
Inhibition of HRV-74, 15, 16, 44, 51, 54, 55, 72, 76 and 89 Viral Infectivity (at 100 TCID50) by Elicited Specific Antibodies Raised Against Recombinant Capsid Proteins and Synthetic Peptides Corresponding to the Conserved Region of the RVs Capsid Proteins. Data are shown as mean ± SE.
Discussion
This study explored the use of bioinformatics tools to analyse all expected immunogenic capsid proteins and to identify the highly conserved ones. The most likely antigenic sites within those capsid proteins were considered as vaccine candidates. Since RV is a diverse pathogen with more than 160 confirmed strains, we focused on identifying a suitable vaccine candidate for RV-A species as it was the most predominant group without neglecting the RV-B. Our four capsid proteins (VP1-4) were found to be variable in length and sequence from one strain to another. According to the aa sequences analysis, VP3 was detected to be the most variable protein among other ones within each group, followed by VP1 and VP2, respectively.
In several studies, VP1 protein has been considered as the most important and potential candidate for vaccine development (19,24-26). In fact, it is the most surface-exposed and immunodominant protein among four capsid proteins and contains most of the motifs known to interact with the cellular receptor ICAM-1 (27,28). In this study, however, it was confirmed that the amino acid sequences of VP1 within individual groups were not well-conserved with an average identity of 70.61% and 76.27% for RV-A and –B, respectively. To further confirm the accuracy of the analysis, data generated for VP1 sequences in this study were compared to those from the study by Ledford et al (14); and the results from two studies were found to be largely consistent, except for the lowest identities between the pairs which were found to be 57.24% (HRV-45 and HRV-49) and 66.9% (HRV-14 and -84) for both groups in our study, respectively. On the other hand, VP2 was discovered to be the most conserved surface-exposed protein with 130 fully conserved residues (48.14% of the full length) at different positions across all RV-A. This was in agreement with the reports from previous studies (29,30). However, it was different from one strain to another and varied in sequence length ranging from 259 to 267 aa residues. The high degree of identity could have been due to the recombination that potentially happened as explained previously (29). Therefore, it was strongly suggested that VP2 be considered in vaccination approach because anti-peptides derived from the VP2 showed neutralizing activity and had been reported to play a special role in the future of RV vaccination (31,32).
As for numbers, VP3 was more conserved than VP1, but without any well-conserved motifs with 9 residues or more were identified. This feature allowed its categorisation (VP3) as the most variable protein among four studied proteins. Similar to the limited analysis performed for examining bioinformatics, limited studies have been carried out for explaining the role of VP3 in immunogenicity. However, VP4 has been discovered to be the most conserved protein compared to other proteins. This finding is in agreement with that of other studies carried out previously (29,30). Within the group, the level of VP4 aa had the greatest degree of identity, ranging from a low level of 88.41% to a higher level of 100% (337 serotype pairs). Despite the fact that VP4 is located inside the virion, the bioinformatics findings have strongly suggested that VP4 may be considered as an immunogenic particle, as supported in a previous study (24). Anti-synthetic peptide antisera have also shown a significant reactivity in vitro. The synthetic peptides corresponding to VP4 have elicited strong cross-neutralizing antibodies against distinct RV stereotypes. These peptides were the longest peptides synthesised in our study, covering the fully conserved region across all RV-A VP4 and representing 72% of the full protein length based on the Bioinformatics conducted in the current study. VP4 is involved in the encoding process which allows the viral RNA to be released into the host cytoplasm (34-37). Neutralizing the viral infectivity by antibody response to VP4 is thought to be due to blocking such a viral site by an antibody which probably prevents the viral genome to be translated to the host cytoplasm and consequently viral infectivity. Since vaccines only need to elicit antibodies that bind to the authentic pathogen with high affinity and work synergistically with other immune system components (24), the induced neutralizing antibodies in this study could have been the best choice to play such a role against the common cold infections. T cells are able to recognise internal viral antigens which are more highly conserved than the surface protein containing neutralizing immunogenic sites (38). As for RVs, immunization with peptides derived from the capsid proteins has been suggested to be capable of inducing cross-serotype reactive T cells against a wide variety of viral serotypes (31).
Conclusions
In conclusion, nine highly conserved regions with the minimum length of 9-mers were identified and characterized within the VP1, VP2, and VP4 protein sequences. These conserved regions across the majority of RV-A may have represented potential immune targets. Remarkably, these conserved regions showed stability over the entire history of RV-A sequences deposited in the NCBI database, as illustrated by their low entropy values and variant frequencies. The synthetic versions of these targets could have been a possible alternative approach to develop the vaccine of choice. Therefore, it was recommended that reverse vaccinology be adopted as a new alternative when conducting RV vaccinology researches due to its speed and accuracy in vaccine screening and designing.
Conflict of Interests
None to declare.
References
- Brooks G. Mycoplasmas & cell wall-defective bacteria. In: Brooks GF, Carroll KC, Butel JS, Morsa SA, Mietzner TA, eds. Jawetz, Melnick & Adelberg’s Medical Microbiology. New York: McGraw-Hill Companies, Inc; 2010.
- Kennedy JL, Pham S, Borish L. Rhinovirus and asthma exacerbations. Immunol Allergy Clin North Am 2019; 39(3):335-44. doi: 10.1016/j.iac.2019.03.003 [Crossref] [ Google Scholar]
- Gern JE. Rhinoviruses and the onset of asthma. In: Bartlett N, Wark P, Knight D, eds. Rhinovirus Infections. Academic Press; 2019. p. 121-36. 10.1016/b978-0-12-816417-4.00005-6
- Han M, Rajput C, Hershenson MB. Rhinovirus attributes that contribute to asthma development. Immunol Allergy Clin North Am 2019; 39(3):345-59. doi: 10.1016/j.iac.2019.03.004 [Crossref] [ Google Scholar]
- O’Neill MB, Laval G, Teixeira JC, Palmenberg AC, Pepperell CS. Genetic susceptibility to severe childhood asthma and rhinovirus-C maintained by balancing selection in humans for 150 000 years. Hum Mol Genet 2020; 29(5):736-44. doi: 10.1093/hmg/ddz304 [Crossref] [ Google Scholar]
- Mallia P, Message SD, Gielen V, Contoli M, Gray K, Kebadze T. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am J Respir Crit Care Med 2011; 183(6):734-42. doi: 10.1164/rccm.201006-0833OC [Crossref] [ Google Scholar]
- Broberg E, Niemelä J, Lahti E, Hyypiä T, Ruuskanen O, Waris M. Human rhinovirus C--associated severe pneumonia in a neonate. J Clin Virol 2011; 51(1):79-82. doi: 10.1016/j.jcv.2011.01.018 [Crossref] [ Google Scholar]
- Renwick N, Schweiger B, Kapoor V, Liu Z, Villari J, Bullmann R. A recently identified rhinovirus genotype is associated with severe respiratory-tract infection in children in Germany. J Infect Dis 2007; 196(12):1754-60. doi: 10.1086/524312 [Crossref] [ Google Scholar]
- Choi EH, Lee HJ, Kim SJ, Eun BW, Kim NH, Lee JA. The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000-2005. Clin Infect Dis 2006; 43(5):585-92. doi: 10.1086/506350 [Crossref] [ Google Scholar]
- Chantzi FM, Papadopoulos NG, Bairamis T, Tsiakou M, Bournousouzis N, Constantopoulos AG. Human rhinoviruses in otitis media with effusion. Pediatr Allergy Immunol 2006; 17(7):514-8. doi: 10.1111/j.1399-3038.2006.00448.x [Crossref] [ Google Scholar]
- Heymann PW. Developing strategies to treat asthma exacerbations caused by rhinovirus. EBioMedicine 2015; 2(1):11-2. doi: 10.1016/j.ebiom.2014.12.012 [Crossref] [ Google Scholar]
- La Rosa G, Fratini M, Della Libera S, Iaconelli M, Muscillo M. Viral infections acquired indoors through airborne, droplet or contact transmission. Ann Ist Super Sanita 2013; 49(2):124-32. doi: 10.4415/ann_13_02_03 [Crossref] [ Google Scholar]
- Blomqvist S, Roivainen M, Puhakka T, Kleemola M, Hovi T. Virological and serological analysis of rhinovirus infections during the first two years of life in a cohort of children. J Med Virol 2002; 66(2):263-8. doi: 10.1002/jmv.2140 [Crossref] [ Google Scholar]
- Ledford RM, Patel NR, Demenczuk TM, Watanyar A, Herbertz T, Collett MS. VP1 sequencing of all human rhinovirus serotypes: insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J Virol 2004; 78(7):3663-74. doi: 10.1128/jvi.78.7.3663-3674.2004 [Crossref] [ Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 1999; 41(2):95-8. [ Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32(5):1792-7. doi: 10.1093/nar/gkh340 [Crossref] [ Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 2011; 28(10):2731-9. doi: 10.1093/molbev/msr121 [Crossref] [ Google Scholar]
- Kiang D, Yagi S, Kantardjieff KA, Kim EJ, Louie JK, Schnurr DP. Molecular characterization of a variant rhinovirus from an outbreak associated with uncommonly high mortality. J Clin Virol 2007; 38(3):227-37. doi: 10.1016/j.jcv.2006.12.016 [Crossref] [ Google Scholar]
- Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng 2005; 99(4):303-10. doi: 10.1263/jbb.99.303 [Crossref] [ Google Scholar]
- Fling SP, Gregerson DS. Peptide and protein molecular weight determination by electrophoresis using a high-molarity tris buffer system without urea. Anal Biochem 1986; 155(1):83-8. doi: 10.1016/0003-2697(86)90228-9 [Crossref] [ Google Scholar]
- Gagnon MC, Williams M, Doucet A, Beauregard M. Replacement of tyr62 by trp in the designer protein milk bundle-1 results in significant improvement of conformational stability. FEBS Lett 2000; 484(2):144-8. doi: 10.1016/s0014-5793(00)02142-6 [Crossref] [ Google Scholar]
- Edlmayr J, Niespodziana K, Popow-Kraupp T, Krzyzanek V, Focke-Tejkl M, Blaas D. Antibodies induced with recombinant VP1 from human rhinovirus exhibit cross-neutralisation. Eur Respir J 2011; 37(1):44-52. doi: 10.1183/09031936.00149109 [Crossref] [ Google Scholar]
- Hu WG, Yin J, Chau D, Hu CC, Lillico D, Yu J. Conformation-dependent high-affinity potent ricin-neutralizing monoclonal antibodies. Biomed Res Int 2013; 2013:471346. doi: 10.1155/2013/471346 [Crossref] [ Google Scholar]
- Katpally U, Fu TM, Freed DC, Casimiro DR, Smith TJ. Antibodies to the buried N terminus of rhinovirus VP4 exhibit cross-serotypic neutralization. J Virol 2009; 83(14):7040-8. doi: 10.1128/jvi.00557-09 [Crossref] [ Google Scholar]
- McCray J, Werner G. Different rhinovirus serotypes neutralized by antipeptide antibodies. Nature 1987; 329(6141):736-8. doi: 10.1038/329736a0 [Crossref] [ Google Scholar]
- Verdaguer N, Fita I, Reithmayer M, Moser R, Blaas D. X-ray structure of a minor group human rhinovirus bound to a fragment of its cellular receptor protein. Nat Struct Mol Biol 2004; 11(5):429-34. doi: 10.1038/nsmb753 [Crossref] [ Google Scholar]
- Rossmann MG, Arnold E, Erickson JW, Frankenberger EA, Griffith JP, Hecht HJ. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985; 317(6033):145-53. doi: 10.1038/317145a0 [Crossref] [ Google Scholar]
- Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW. The major human rhinovirus receptor is ICAM-1. Cell 1989; 56(5):839-47. doi: 10.1016/0092-8674(89)90688-0 [Crossref] [ Google Scholar]
- Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009; 324(5923):55-9. doi: 10.1126/science.1165557 [Crossref] [ Google Scholar]
- Glanville N, McLean GR, Guy B, Lecouturier V, Berry C, Girerd Y. Cross-serotype immunity induced by immunization with a conserved rhinovirus capsid protein. PLoS Pathog 2013; 9(9):e1003669. doi: 10.1371/journal.ppat.1003669 [Crossref] [ Google Scholar]
- Hastings GZ, Speller SA, Francis MJ. Neutralizing antibodies to human rhinovirus produced in laboratory animals and humans that recognize a linear sequence from VP2. J Gen Virol 1990; 71(Pt 12):3055-9. doi: 10.1099/0022-1317-71-12-3055 [Crossref] [ Google Scholar]
- Skern T, Neubauer C, Frasel L, Gründler P, Sommergruber W, Zorn M. A neutralizing epitope on human rhinovirus type 2 includes amino acid residues between 153 and 164 of virus capsid protein VP2. J Gen Virol 1987; 68(Pt 2):315-23. doi: 10.1099/0022-1317-68-2-315 [Crossref] [ Google Scholar]
- Tormo J, Blaas D, Parry NR, Rowlands D, Stuart D, Fita I. Crystal structure of a human rhinovirus neutralizing antibody complexed with a peptide derived from viral capsid protein VP2. EMBO J 1994; 13(10):2247-56. [ Google Scholar]
- Seechurn P, Knowles NJ, McCauley JW. The complete nucleotide sequence of a pathogenic swine vesicular disease virus. Virus Res 1990; 16(3):255-74. doi: 10.1016/0168-1702(90)90052-d [Crossref] [ Google Scholar]
- Tuthill TJ, Bubeck D, Rowlands DJ, Hogle JM. Characterization of early steps in the poliovirus infection process: receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J Virol 2006; 80(1):172-80. doi: 10.1128/jvi.80.1.172-180.2006 [Crossref] [ Google Scholar]
- Davis MP, Bottley G, Beales LP, Killington RA, Rowlands DJ, Tuthill TJ. Recombinant VP4 of human rhinovirus induces permeability in model membranes. J Virol 2008; 82(8):4169-74. doi: 10.1128/jvi.01070-07 [Crossref] [ Google Scholar]
- Danthi P, Tosteson M, Li QH, Chow M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J Virol 2003; 77(9):5266-74. doi: 10.1128/jvi.77.9.5266-5274.2003 [Crossref] [ Google Scholar]
- Gilbert SC. T-cell-inducing vaccines - what’s the future. Immunology 2012; 135(1):19-26. doi: 10.1111/j.1365-2567.2011.03517.x [Crossref] [ Google Scholar]